一種甘薯多糖的應用的制作方法
本發明涉及甘薯多糖的應用。
背景技術:
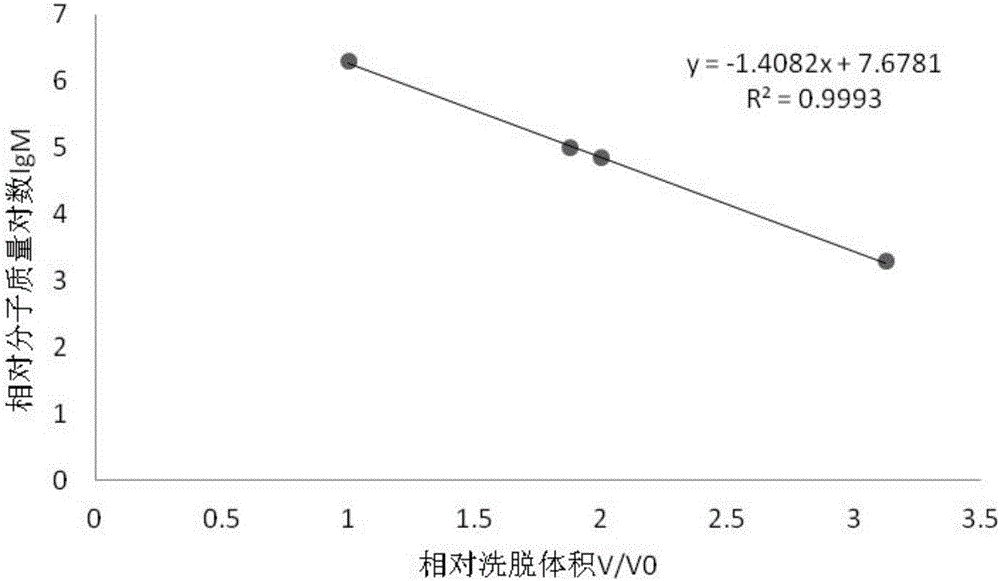
:三-(2,3-二溴丙基)異氰酸酯(Tris-(2,3-dibromopropyl)isocyanurate,TBC)是一種含有三嗪環結構的溴系化合物,在國內外廣泛用于阻燃劑,聚苯乙烯、聚烯烴、不飽和聚酯、聚氨酯等塑料、紡織品和橡膠等化工產品的合成。據不完全統計,自從1991年起,我國的TBC總產量不低于500噸/年,且呈現逐年增長的趨勢;此外,TBC在歐洲、美國和日本等發達國家的應用也越來越廣泛。2009年,我國學者首次在渤海中檢出TBC(0.19-2.64ng/g),研究結果還顯示TBC具有生物蓄積性,對環境可能會產生持久性污染。人們發現,TBC對斑馬魚和嚙齒類動物的肝臟具有很強的損傷作用。隨著TBC用量的不斷增加,人群不可避免地通過直接或間接的途徑與其發生接觸,因此針對TBC對肝臟毒性作用的預防和治療具有重要的戰略意義和研究價值。技術實現要素:本發明的目的是為了解決由TBC引起的肝損傷的問題,而提供一種甘薯多糖的應用。本發明所述甘薯多糖作為制備治療三-(2,3-二溴丙基)異氰酸酯致肝損傷的藥物應用。所述甘薯多糖為由煙薯25提取的煙薯多糖。所述煙薯多糖為具有α-1,3-葡聚糖鏈的煙薯多糖,其重復結構單元中含有1→3和1→6兩種糖苷鍵,主鏈為1→3糖苷鍵,側鏈由1→6糖苷鍵組成,糖殘基為α構型,分支度為0.33,其結構式為:其中n為340~345,相對分子質量為2.40×105Da~2.46×105Da。所述具有α-1,3-葡聚糖鏈的煙薯多糖的制備方法為:一、制備煙薯粗多糖:將煙薯25清洗干凈,去皮,切塊,自然晾干,粉碎,過50目篩,得煙薯粉末,將煙薯粉末放入質量分數為95%的乙醇中浸泡24h~48h進行脫脂,取出后自然晾干,然后進行熱水浸提,抽濾得提取液,將提取液進行減壓濃縮,得到濃縮液I,將濃縮液I放入質量分數為95%的乙醇溶液中浸泡12h~24h,抽濾得沉淀I,將沉淀I真空干燥后得到沉淀粉末,將沉淀粉末放入蒸餾水中,得到濃度為2mg/mL的粗多糖溶液I,然后用Sevag法脫蛋白,取上清液,將上清液進行減壓濃縮,得到濃縮液II,將濃縮液II放入質量分數為95%的乙醇溶液中浸泡12h~24h,抽濾得沉淀II,然后將沉淀II進行真空干燥,得到煙薯粗多糖;所述的煙薯粉末的質量與質量分數為95%的乙醇的體積比為1g:(5mL~10mL);所述的將濃縮液I放入質量分數為95%的乙醇溶液中浸泡12h~24h中的濃縮液I與質量分數為95%的乙醇溶液的體積比為1:(3~4);所述的將濃縮液II放入質量分數為95%的乙醇溶液中浸泡12h~24h中的濃縮液II與質量分數為95%的乙醇溶液的體積比為1:(3~4);二、純化:將步驟一得到的煙薯粗多糖放入蒸餾水中,用0.45μm的微孔濾膜過濾,得到濃度為2mg/mL的粗多糖溶液II,用DEAE-52纖維素層析柱進行初步分離,得到分離后的洗脫液I,將分離后的洗脫液I依次進行流水透析1天,蒸餾水透析2天,再進行減壓濃縮和真空冷凍干燥,得到煙薯多糖組分,將煙薯多糖組分放入蒸餾水中,用0.45μm的微孔濾膜過濾,得到濃度為2mg/mL的煙薯多糖組分溶液,然后用SephadexG-200層析柱對煙薯多糖組分溶液進行分離純化,得到分離后的洗脫液II,將分離后的洗脫液II進行減壓濃縮和真空冷凍干燥,得到具有α-1,3-葡聚糖鏈的煙薯多糖;步驟二中所述的流水透析的具體步驟為:將洗脫液I裝入截留分子量為8000道爾頓~11000道爾頓的透析袋中,將透析袋置于1000mL的燒杯中,室溫下流水持續浸泡透析1天;步驟二所述的蒸餾水透析的具體步驟為:將流水透析處理后的透析袋置于盛有1000mL蒸餾水的燒杯中,蒸餾水浸泡透析2天,其中每6h~12h更換1次蒸餾水;所述的DEAE-52纖維素層析柱進行初步分離的條件為:進行氯化鈉溶液梯度洗脫,所述的氯化鈉溶液濃度依次為0.5mol/L和1.0mol/L,每次所用氯化鈉溶液的體積為DEAE-52纖維素層析柱體積的1.5倍~2倍,恒流泵控制洗脫流速為1mL/min,粗多糖溶液II的上樣量為2mL,用自動部分收集器每3mL收集1管,收集第20管至第60管的洗脫液為分離后的洗脫液I;所述的DEAE-52纖維素層析柱內徑為2.6cm,長度為55cm;所述的SephadexG-200層析柱對煙薯粗多糖組分溶液進行分離純化的條件為:洗脫液為蒸餾水,所用蒸餾水的體積為SephadexG-200層析柱體積的1.5倍~2倍,恒流泵控制洗脫的流速為0.5mL/min,煙薯粗多糖組分溶液的上樣量為1mL,用自動部分收集器每3mL收集1管,收集第25管至第50管的洗脫液為分離后的洗脫液II;所述的SephadexG-200層析柱內徑為1.6cm,長度為80cm。本發明的有益效果:本發明從體外試驗和體內試驗兩個部分進行闡述甘薯多糖用于治療三-(2,3-二溴丙基)異氰酸酯致肝損傷的方法。體外試驗采用Wistar大鼠喂養試驗。體內試驗以HepG2細胞為靶細胞,以TBC【三-(2,3-二溴丙基)異氰酸酯(Tris-(2,3-dibromopropyl)isocyanurate,TBC】誘導HepG2細胞凋亡的劑量處理細胞后,再用不同劑量和不同處理時間來觀察甘薯多糖(purplesweetpotatopolysaccharide,PSPP)對TBC致HepG2細胞凋亡的保護作用。肝功能生化檢測結果顯示,應用甘薯多糖治療三-(2,3-二溴丙基)異氰酸酯致肝損傷后,谷丙轉氨酶、谷草轉氨酶=血清總蛋白、血清白蛋白、尿素氮、肌酐、膽固醇及甘油三酯等相比TBC染毒組均有很大改善。肝功能生化檢測結果顯示,應用甘薯多糖治療三-(2,3-二溴丙基)異氰酸酯致肝損傷后,堿性磷酸酶(ALP),丙氨酸轉氨酶(ALT),天冬氨酸轉氨酶(AST),總蛋白(TP),白蛋白(ALB),總膽紅素(TBIL),總膽汁酸(TBA),總膽固醇(TCHO),三酰甘油(TG),葡萄糖(GLU)相比TBC染毒組均有很大改善。應用甘薯多糖治療三-(2,3-二溴丙基)異氰酸酯致肝損傷后,肝/體比值相比TBC染毒組有明顯的改善病理組織學檢查結果顯示,肝臟出現空泡和炎細胞浸潤,可得出結論應用甘薯多糖治療三-(2,3-二溴丙基)異氰酸酯致肝損傷后,對肝臟有改善和保護作用。對由TBC誘導HepG2細胞凋亡的凋亡率檢測結果顯示,應用甘薯多糖治療三-(2,3-二溴丙基)異氰酸酯致肝損傷后,凋亡率有明顯的降低。本發明的具有α-1,3-葡聚糖鏈的煙薯多糖的多糖含量為98.47%。附圖說明圖1為實施例1步驟一制備的煙薯粗多糖在波長200-400nm區間進行紫外掃描的光譜圖;圖2為實施例1中標準葡聚糖凝膠色譜標準曲線;圖3為實施例1步驟一制備的煙薯粗多糖的紅外掃描圖譜;圖4為實施例1制備的具有α-1,3-葡聚糖鏈的煙薯多糖的甲基化糖殘基氣相色譜圖,峰1對應著甲基化糖殘基1,峰2對應著甲基化糖殘基2,峰3對應著甲基化糖殘基3;圖5是甲基化糖殘基1的質譜圖;圖6是甲基化糖殘基2的質譜圖;圖7是甲基化糖殘基3的質譜圖;圖8為試驗(四)中對照組的肝組織病理學切片(x400)照片;圖9為試驗(四)中100mg/kg·bw劑量實施例1的甘薯多糖干預組的肝組織病理學切片(x400)照片;圖10為試驗(四)中50mg/kg·bw劑量實施例1的甘薯多糖干預組的肝組織病理學切片(x400)照片;圖11為試驗(四)中TBC染毒組的肝組織病理學切片(x400)照片。具體實施方式具體實施方式一:本實施方式所述甘薯多糖作為制備治療三-(2,3-二溴丙基)異氰酸酯致肝損傷的藥物應用。本實施方式體外試驗和體內試驗兩個部分進行闡述甘薯多糖用于治療三-(2,3-二溴丙基)異氰酸酯致肝損傷的方法。體外試驗采用Wistar大鼠喂養試驗。體內試驗以HepG2細胞為靶細胞,以TBC【三-(2,3-二溴丙基)異氰酸酯(Tris-(2,3-dibromopropyl)isocyanurate,TBC】誘導HepG2細胞凋亡的劑量處理細胞后,再用不同劑量和不同處理時間來觀察PSPP對TBC致HepG2細胞凋亡的保護作用。肝功能生化檢測結果顯示,應用甘薯多糖治療三-(2,3-二溴丙基)異氰酸酯致肝損傷后,谷丙轉氨酶、谷草轉氨酶=血清總蛋白、血清白蛋白、尿素氮、肌酐、膽固醇及甘油三酯等相比TBC染毒組均有很大改善。肝功能生化檢測結果顯示,應用甘薯多糖治療三-(2,3-二溴丙基)異氰酸酯致肝損傷后,堿性磷酸酶(ALP),丙氨酸轉氨酶(ALT),天冬氨酸轉氨酶(AST),總蛋白(TP),白蛋白(ALB),總膽紅素(TBIL),總膽汁酸(TBA),總膽固醇(TCHO),三酰甘油(TG),葡萄糖(GLU)相比TBC染毒組均有很大改善。應用甘薯多糖治療三-(2,3-二溴丙基)異氰酸酯致肝損傷后,肝/體比值相比TBC染毒組有明顯的改善病理組織學檢查結果顯示,肝臟出現空泡和炎細胞浸潤,可得出結論應用甘薯多糖治療三-(2,3-二溴丙基)異氰酸酯致肝損傷后,對肝臟有改善和保護作用。對由TBC誘導HepG2細胞凋亡的凋亡率檢測結果顯示,應用甘薯多糖治療三-(2,3-二溴丙基)異氰酸酯致肝損傷后,凋亡率有明顯的降低。具體實施方式二:本實施方式與具體實施方式一不同的是:將所述甘薯多糖制備成靜脈注射劑或口服劑。其他步驟及參數與具體實施方式一相同。具體實施方式三:本實施方式與具體實施方式一或二不同的是:所述口服劑為口服液、膠囊或片劑。其他步驟及參數與具體實施方式一或二相同。具體實施方式四:本實施方式與具體實施方式一至三之一不同的是:所述甘薯多糖為由煙薯25提取的煙薯多糖。其他步驟及參數與具體實施方式一至三之一相同。具體實施方式五:本實施方式與具體實施方式一至四之一不同的是:所述煙薯多糖為具有α-1,3-葡聚糖鏈的煙薯多糖,其重復結構單元中含有1→3和1→6兩種糖苷鍵,主鏈為1→3糖苷鍵,側鏈由1→6糖苷鍵組成,糖殘基為α構型,分支度為0.33,其結構式為:其中n為340~345,相對分子質量為2.40×105Da~2.46×105Da。其他步驟及參數與具體實施方式一至四之一相同。具體實施方式六:本實施方式與具體實施方式一至五之一不同的是:所述具有α-1,3-葡聚糖鏈的煙薯多糖的制備方法為:一、制備煙薯粗多糖:將煙薯25清洗干凈,去皮,切塊,自然晾干,粉碎,過50目篩,得煙薯粉末,將煙薯粉末放入質量分數為95%的乙醇中浸泡24h~48h進行脫脂,取出后自然晾干,然后進行熱水浸提,抽濾得提取液,將提取液進行減壓濃縮,得到濃縮液I,將濃縮液I放入質量分數為95%的乙醇溶液中浸泡12h~24h,抽濾得沉淀I,將沉淀I真空干燥后得到沉淀粉末,將沉淀粉末放入蒸餾水中,得到濃度為2mg/mL的粗多糖溶液I,然后用Sevag法脫蛋白,取上清液,將上清液進行減壓濃縮,得到濃縮液II,將濃縮液II放入質量分數為95%的乙醇溶液中浸泡12h~24h,抽濾得沉淀II,然后將沉淀II進行真空干燥,得到煙薯粗多糖;所述的煙薯粉末的質量與質量分數為95%的乙醇的體積比為1g:(5mL~10mL);所述的將濃縮液I放入質量分數為95%的乙醇溶液中浸泡12h~24h中的濃縮液I與質量分數為95%的乙醇溶液的體積比為1:(3~4);所述的將濃縮液II放入質量分數為95%的乙醇溶液中浸泡12h~24h中的濃縮液II與質量分數為95%的乙醇溶液的體積比為1:(3~4);二、純化:將步驟一得到的煙薯粗多糖放入蒸餾水中,用0.45μm的微孔濾膜過濾,得到濃度為2mg/mL的粗多糖溶液II,用DEAE-52纖維素層析柱進行初步分離,得到分離后的洗脫液I,將分離后的洗脫液I依次進行流水透析1天,蒸餾水透析2天,再進行減壓濃縮和真空冷凍干燥,得到煙薯多糖組分,將煙薯多糖組分放入蒸餾水中,用0.45μm的微孔濾膜過濾,得到濃度為2mg/mL的煙薯多糖組分溶液,然后用SephadexG-200層析柱對煙薯多糖組分溶液進行分離純化,得到分離后的洗脫液II,將分離后的洗脫液II進行減壓濃縮和真空冷凍干燥,得到具有α-1,3-葡聚糖鏈的煙薯多糖;步驟二中所述的流水透析的具體步驟為:將洗脫液I裝入截留分子量為8000道爾頓~11000道爾頓的透析袋中,將透析袋置于1000mL的燒杯中,室溫下流水持續浸泡透析1天;步驟二所述的蒸餾水透析的具體步驟為:將流水透析處理后的透析袋置于盛有1000mL蒸餾水的燒杯中,蒸餾水浸泡透析2天,其中每6h~12h更換1次蒸餾水;所述的DEAE-52纖維素層析柱進行初步分離的條件為:進行氯化鈉溶液梯度洗脫,所述的氯化鈉溶液濃度依次為0.5mol/L和1.0mol/L,每次所用氯化鈉溶液的體積為DEAE-52纖維素層析柱體積的1.5倍~2倍,恒流泵控制洗脫流速為1mL/min,粗多糖溶液II的上樣量為2mL,用自動部分收集器每3mL收集1管,收集第20管至第60管的洗脫液為分離后的洗脫液I;所述的DEAE-52纖維素層析柱內徑為2.6cm,長度為55cm;所述的SephadexG-200層析柱對煙薯粗多糖組分溶液進行分離純化的條件為:洗脫液為蒸餾水,所用蒸餾水的體積為SephadexG-200層析柱體積的1.5倍~2倍,恒流泵控制洗脫的流速為0.5mL/min,煙薯粗多糖組分溶液的上樣量為1mL,用自動部分收集器每3mL收集1管,收集第25管至第50管的洗脫液為分離后的洗脫液II;所述的SephadexG-200層析柱內徑為1.6cm,長度為80cm。其他步驟及參數與具體實施方式一至五之一相同。本實施方式的具有α-1,3-葡聚糖鏈的煙薯多糖的多糖含量為98.47%。具體實施方式七:本實施方式與具體實施方式六不同的是:步驟一中所述的熱水浸提的條件為:提取溫度為65℃~85℃,提取時間1.5h~3h,所述的自然晾干后的煙薯25與水的質量比為1:(10~20),提取次數為2次~4次。其他步驟及參數與具體實施方式六相同。具體實施方式八:本實施方式與具體實施方式六和七不同的是:步驟一所述的減壓濃縮的過程為:將提取液放入旋轉蒸發瓶中,在壓強為0.05MPa~0.08MPa和水浴溫度為55℃~65℃的條件下將提取液壓縮至原體積的15%~30%。其他步驟及參數與具體實施方式六和七相同。具體實施方式九:本實施方式與具體實施方式六至八之一不同的是:步驟一所述的Sevag法脫蛋白的過程為:將濃度為2mg/mL的粗多糖溶液I與Sevag液等體積混合,震蕩充分混勻,然后在轉速為3000r·min-1離心10min,取上清液;將上清液重復Sevag法脫蛋白的過程5次~10次。其他步驟及參數與具體實施方式六至八之一相同。具體實施方式十:本實施方式與具體實施方式六至九之一不同的是:步驟一中所述的將沉淀I真空干燥的過程為:將沉淀I裝入研缽中,將研缽置于真空干燥箱中,干燥溫度為50℃~70℃,壓強為0.02MPa~0.05MPa,干燥時間為12h~24h;步驟一中所述的將沉淀II真空干燥的過程為:將沉淀II裝入研缽中,將研缽置于真空干燥箱中,干燥溫度為50℃~70℃,壓強為0.02MPa~0.05MPa,干燥時間為12h~24h。其他步驟及參數與具體實施方式六至九之一相同。具體實施方式十一:本實施方式與具體實施方式六至十之一不同的是:步驟二中所述的將分離后的洗脫液I依次進行減壓濃縮和真空冷凍干燥中的減壓濃縮步驟為:將分離后的洗脫液I放入旋轉蒸發瓶中,在壓強為0.05MPa~0.08MPa和水浴溫度為55℃~65℃的條件下將洗脫液I壓縮至原體積的15%~30%,得濃縮液;真空冷凍干燥的過程為:將濃縮液裝入2mL的安瓿瓶中,將安瓿瓶置于真空冷凍干燥機中,設定冷凍干燥制品溫度為-25℃~-40℃,冷凍時間為12h~24h,壓力5Pa~40Pa,解析溫度為20℃~40℃,解析時間為8h~18h。其他步驟及參數與具體實施方式六至十之一相同。具體實施方式十二:本實施方式與具體實施方式六至十一之一不同的是:步驟二中所述的將分離后的洗脫液II依次進行減壓濃縮和真空冷凍干燥中的減壓濃縮步驟為:將分離后的洗脫液II放入旋轉蒸發瓶中,在壓強為0.05MPa~0.08MPa和水浴溫度為55℃~65℃的條件下將洗脫液II壓縮至原體積的15%~30%,得濃縮液;真空冷凍干燥的過程為:將濃縮液裝入2mL的安瓿瓶中,將安瓿瓶置于真空冷凍干燥機中,設定冷凍干燥制品溫度為-25℃~-40℃,冷凍時間為12h~24h,壓力5Pa~40Pa,解析溫度為20℃~40℃,解析時間為8h~18h。其他步驟及參數與具體實施方式六至十一之一相同。用以下試驗來驗證本發明的效果實施例1:本實施例具有α-1,3-葡聚糖鏈的煙薯多糖(以下簡稱為PSPP)的制備方法具體為:一、制備煙薯粗多糖:將煙薯25清洗干凈,去皮,切塊,自然晾干,粉碎,過50目篩,得煙薯粉末,將煙薯粉末放入質量分數為95%的乙醇中浸泡48h進行脫脂,取出后自然晾干,然后進行熱水浸提,抽濾得提取液,將提取液進行減壓濃縮,得到濃縮液I,將濃縮液I放入質量分數為95%的乙醇溶液中浸泡24h,抽濾得沉淀I,將沉淀I真空干燥后得到沉淀粉末,將沉淀粉末放入蒸餾水中,得到濃度為2mg/mL的粗多糖溶液I,然后用Sevag法脫蛋白,取上清液,將上清液進行減壓濃縮,得到濃縮液II,將濃縮液II放入質量分數為95%的乙醇溶液中浸泡24h,抽濾得沉淀II,然后將沉淀II進行真空干燥,得到煙薯粗多糖;所述的煙薯粉末的質量與質量分數為95%的乙醇的體積比為1g:5mL;所述的將濃縮液I放入質量分數為95%的乙醇溶液中浸泡24h中的濃縮液I與質量分數為95%的乙醇溶液的體積比為1:3;所述的將濃縮液II放入質量分數為95%的乙醇溶液中浸泡24h中的濃縮液II與質量分數為95%的乙醇溶液的體積比為1:3;二、純化:將步驟一得到的煙薯粗多糖放入蒸餾水中,用0.45μm的微孔濾膜過濾,得到濃度為2mg/mL的粗多糖溶液II,用DEAE-52纖維素層析柱進行初步分離,得到分離后的洗脫液I,將分離后的洗脫液I依次進行流水透析1天,蒸餾水透析2天,再進行減壓濃縮和真空冷凍干燥,得到煙薯多糖組分,將煙薯多糖組分放入蒸餾水中,用0.45μm的微孔濾膜過濾,得到濃度為2mg/mL的煙薯多糖組分溶液,然后用SephadexG-200層析柱對煙薯多糖組分溶液進行分離純化,得到分離后的洗脫液II,將分離后的洗脫液II進行減壓濃縮和真空冷凍干燥,得到具有α-1,3-葡聚糖鏈的煙薯多糖;步驟二中所述的流水透析的具體步驟為:將洗脫液I裝入截留分子量為8000道爾頓~11000道爾頓的透析袋中,將透析袋置于1000mL的燒杯中,室溫下流水持續浸泡透析1天;步驟二所述的蒸餾水透析的具體步驟為:將流水透析處理后的透析袋置于盛有1000mL蒸餾水的燒杯中,蒸餾水浸泡透析2天,其中每12h更換1次蒸餾水;所述的DEAE-52纖維素層析柱進行初步分離的條件為:進行氯化鈉溶液梯度洗脫,所述的氯化鈉溶液濃度依次為0.5mol/L和1.0mol/L,每次所用氯化鈉溶液的體積為DEAE-52纖維素層析柱體積的1.5倍~2倍,恒流泵控制洗脫流速為1mL/min,粗多糖溶液II的上樣量為2mL,用自動部分收集器每3mL收集1管,收集第20管至第60管的洗脫液為分離后的洗脫液I;所述的DEAE-52纖維素層析柱內徑為2.6cm,長度為55cm;所述的SephadexG-200層析柱對煙薯粗多糖組分溶液進行分離純化的條件為:洗脫液為蒸餾水,所用蒸餾水的體積為SephadexG-200層析柱體積的1.5倍~2倍,恒流泵控制洗脫的流速為0.5mL/min,煙薯粗多糖組分溶液的上樣量為1mL,用自動部分收集器每3mL收集1管,收集第25管至第50管的洗脫液為分離后的洗脫液II;所述的SephadexG-200層析柱內徑為1.6cm,長度為80cm。步驟一中所述的熱水浸提的條件為:提取溫度為80℃,提取時間2h,所述的自然晾干后的煙薯25與水的質量比為1:10,提取次數為3次。步驟一所述的減壓濃縮的過程為:將提取液放入旋轉蒸發瓶中,在壓強為0.07MPa和水浴溫度為60℃的條件下將提取液壓縮至原體積的20%。步驟一所述的Sevag法脫蛋白的過程為:將濃度為2mg/mL的粗多糖溶液I與Sevag液等體積混合,震蕩充分混勻,然后在轉速為3000r·min-1離心10min,取上清液;將上清液重復Sevag法脫蛋白的過程8次。步驟一中所述的將沉淀I真空干燥的過程為:將沉淀I裝入研缽中,將研缽置于真空干燥箱中,干燥溫度為60℃,壓強為0.03MPa,干燥時間為12h;步驟一中所述的將沉淀II真空干燥的過程為:將沉淀II裝入研缽中,將研缽置于真空干燥箱中,干燥溫度為60℃,壓強為0.03MPa,干燥時間為12h。步驟二中所述的將分離后的洗脫液I依次進行減壓濃縮和真空冷凍干燥中的減壓濃縮步驟為:將分離后的洗脫液I放入旋轉蒸發瓶中,在壓強為0.07MPa和水浴溫度為60℃的條件下將洗脫液I壓縮至原體積的20%,得濃縮液;真空冷凍干燥的過程為:將濃縮液裝入2mL的安瓿瓶中,將安瓿瓶置于真空冷凍干燥機中,設定冷凍干燥制品溫度為-40℃,冷凍時間為24h,壓力20Pa,解析溫度為35℃,解析時間為12h。步驟二中所述的將分離后的洗脫液II依次進行減壓濃縮和真空冷凍干燥中的減壓濃縮步驟為:將分離后的洗脫液II放入旋轉蒸發瓶中,在壓強為0.07MPa和水浴溫度為60℃的條件下將洗脫液II壓縮至原體積的20%,得濃縮液;真空冷凍干燥的過程為:將濃縮液裝入2mL的安瓿瓶中,將安瓿瓶置于真空冷凍干燥機中,設定冷凍干燥制品溫度為-40℃,冷凍時間為24h,壓力20Pa,解析溫度為35℃,解析時間為12h。對實施例1制備的具有α-1,3-葡聚糖鏈的煙薯多糖(PSPP)的表征檢測:一、煙薯粗多糖紫外光譜掃描:對實施例1步驟一制備的煙薯粗多糖在波長200-400nm區間進行紫外掃描,掃描圖譜如圖1所示,由光譜圖可以看出,在260-280nm之間沒有明顯的特征峰,說明實施例1步驟一制備的煙薯粗多糖中不含有核酸和蛋白質成分,脫蛋白完全,初步純化后對實施例1步驟一制備的煙薯粗多糖利用苯酚硫酸法檢測計算得到多糖含量為70.73%。二、實施例1制備的具有α-1,3-葡聚糖鏈的煙薯多糖相對分子量的測定:1、多糖相對分子質量標準曲線制作精密稱取藍色葡聚糖T20、T70、T100、T2000各50mg,分別定容于100mL容量瓶中,濃度均為0.5mg/mL,分別取1mL,上樣于SephadexG-200層析柱,用恒流泵泵入單蒸水洗脫,用部分自動收集器每2mL/管收集,苯酚-硫酸跟蹤檢測,收集合并標準糖液完全洗脫時體積V。分別記為V1、V2、V3和V0。以V/V0為橫坐標,相對分子質量對數lgM為縱坐標繪制葡聚糖凝膠色譜標準曲線,計算回歸方程。收集完全洗脫已知分子量的藍色葡聚糖T20、T70、T100和T2000分別為25mL、16mL、15mL和8mL。縱坐標由已知分子量的藍色葡聚糖lgM分別為3.30103、4.845098、5、6.30103。橫坐標由V/V0分別為3.125、2、1.25、1。計算線性回歸方程為y=-1.4082x+7.6781,相關系數R2=0.9993。表1標準葡聚糖凝膠色譜數據測定圖2為標準葡聚糖凝膠色譜標準曲線。2、樣品相對分子質量測定精密稱取實施例1制備的具有α-1,3-葡聚糖鏈的煙薯多糖50mg,定容于100mL容量瓶中,濃度為0.5mg/mL,按照步驟1的方法上樣,洗脫,計算完全洗脫時體積V利用標準曲線分別計算對應的相對分子質量,得到洗脫體積分別為13mL,經回歸方程計算驗一制備的具有α-1,3-葡聚糖鏈的煙薯多糖的相對分子質量為2.45×105Da。三、實施例1制備的具有α-1,3-葡聚糖鏈的煙薯多糖紅外光譜掃描取1mg的實施例1制備的具有α-1,3-葡聚糖鏈的煙薯多糖干燥粉末,與200mgKBr粉末混勻,在瑪瑙研缽中研磨,小心將其置于濾紙上,用壓片機壓成薄片,然后上傅立葉紅外光譜儀測定,在4000-400cm-1區間進行紅外掃描,掃描圖譜如圖3。由圖3可知,在3200cm-1-2800cm-1和1400cm-1-1200cm-1,實施例1制備的具有α-1,3-葡聚糖鏈的煙薯多糖有特征吸收峰,說明實施例1制備的具有α-1,3-葡聚糖鏈的煙薯多糖具有糖類特征,在960cm-1-730cm-1為多糖特征吸收峰,3604cm-1有O-H伸縮振動的強吸收峰,1773cm-1是-CHO或C=O的伸縮振動峰,2860cm-1有C-H伸縮振動峰,在1033cm-1有特征吸收峰說明實施例1制備的具有α-1,3-葡聚糖鏈的煙薯多糖為吡喃型糖環,在833cm-1有吸收峰,證明實施例1制備的具有α-1,3-葡聚糖鏈的煙薯多糖糖苷鍵構型含有α-糖苷鍵。四、實施例1制備的具有α-1,3-葡聚糖鏈的煙薯多糖甲基化分析取干燥的實施例1制備的具有α-1,3-葡聚糖鏈的煙薯多糖0.1mg于反應管→溶于0.3mL無水DMSO→充入氮氣,超聲水浴30min→加入10mg氫氧化鈉粉末→充入氮氣,超聲水浴20min→冰浴至樣品溶液凝固→充入氮氣,加0.1mL碘甲烷→封住反應1h→充入氮氣→加單蒸水0.2mL→加氯仿萃取2次,每次1mL→合并氯仿液,加入無水硫酸鈉,過夜→過濾除去無水硫酸鈉,減壓蒸干氯仿→得到甲基化產物→加入2mol/L三氟乙酸0.5mL,100℃反應2h→減壓蒸干,加甲醇0.1mL,充入氮氣→加入5mg硼氫化鈉,2mL單蒸水,反應2h→滴加1mol/L乙酸中和→加入0.1mL甲醇,蒸干→加入無水吡啶和無水醋酸酐各0.5mL,反應2h→減壓蒸干→加0.5mL氯仿溶解→進行GC-MS分析。GC-MS分析條件:色譜柱為EC-1column,填充柱0.25mm×25m,程序升溫100~250℃,載氣為氮氣,EI源70eV,進樣量1μL。圖4為實施例1制備的具有α-1,3-葡聚糖鏈的煙薯多糖的甲基化糖殘基氣相色譜圖,可以看出有三個峰,分別對應著三個甲基化糖殘基1、2和3,圖5、6和7是三個甲基化糖殘基的質譜圖,可以看出三個糖殘基碎片分別是甲基化糖殘基1:2,3,4,6-Me4-Glc、甲基化糖殘基2:2,4-Me2-Glc和甲基化糖殘基3:2,4,6-Me3-Glc,推測出實施例1制備的具有α-1,3-葡聚糖鏈的煙薯多糖的連接方式為1-Glc、1,3,6-Glc和1,3-Glc。由圖4可知,甲基化糖殘基2和3的摩爾比為23.84和15.06可以推斷出為實施例1制備的具有α-1,3-葡聚糖鏈的煙薯多糖的的主鏈,而摩爾比4.58甲基化糖殘基1值小為支鏈組成。實施例1制備的具有α-1,3-葡聚糖鏈的煙薯多糖的結構:主鏈由1→3和1→3→6兩種α-葡聚糖交替構成,在主鏈部分6位上有支鏈,由1-Glc構成。所以,實施例1制備的具有α-1,3-葡聚糖鏈的煙薯多糖的結構式:n為340。表2甲基化分析結果對實施例1制備的具有α-1,3-葡聚糖鏈的煙薯多糖(以下簡稱為PSPP)干預TBC致肝損傷的效果檢測:實施例1制備的具有α-1,3-葡聚糖鏈的煙薯多糖(PSPP)干預TBC致肝損傷的體外喂養Wistar大鼠試驗過程如下:將體重為60g~70g的Wistar大鼠隨機分成四組,每組20只,雌雄各半,四組分別記為陰性對照組、陽性對照組(TBC染毒組)、PSPP低劑量干預組和PSPP高劑量干預組,其中四組所做處理如下:陰性對照組:每天飼喂普通飼料,自由攝食;陽性對照組(TBC染毒組):每天以TBC灌胃進行染毒,TBC的劑量以大鼠體重計為0.625g/kg,飼喂普通飼料,自由攝食;PSPP低劑量干預組:每天先以TBC灌胃進行染毒,TBC的劑量以大鼠體重計為0.625g/kg,染毒后飼喂摻入PSPP的普通飼料,PSPP的劑量每天以Wistar大鼠體重計為50mg/kg,自由攝食;PSPP高劑量干預組:每天先以TBC灌胃進行染毒,TBC的劑量以大鼠體重計為0.625g/kg,染毒后飼喂摻入PSPP的普通飼料,PSPP的劑量每天以Wistar大鼠體重計為100mg/kg,自由攝食;試驗過程中采用用酪蛋白調整各組動物飼料中蛋白含量一致,試驗連續三十天。(一)對生長情況的監測與結果:在三十天的喂養過程中,每周測量一次體重,計算一次體重增重。在飲食攝取量方面,稱量灑食量和剩余量,計算一次攝食量。得到如表3所示的喂養試驗結果,從表3中可以看出TBC染毒組、PSPP低劑量干預組(50mg/kg·bw;bodyweight,bw)、PSPP高劑量干預組(100mg/kg·bw;bodyweight,bw)和陰性對照組的體重增長呈現出明顯的梯度關系,雄性組和雌性組的兩個劑量干預組都與TBC染毒組具有明顯的差異,且存在統計學差異。表3三十天喂養試驗結果注:﹡與對照組比較,p<0.05.(二)肝功能生化檢測及結果:三十天的喂養結束后,動物麻醉條件下腹主動脈取血分離血清,采用生化自動分析儀測定肝功能生化指標,得到如表4和表5所示的血液生化指標檢測結果。①表4給出了谷丙轉氨酶、谷草轉氨酶(包括中劑量組)、血清總蛋白、膽固醇及甘油三酯的檢測結果,從表4可以看出,100mg/kg·bwPSPP干預組雄性大鼠谷丙轉氨酶、谷草轉氨酶、血清總蛋白、膽固醇及甘油三酯均相對TBC染毒組有極大的改善,且存在統計學意義。PSPP兩個劑量的干預組雌性大鼠的谷草轉氨酶及100mg/kg·bwPSPP干預組的血清總蛋白、血清白蛋白、尿素氮、肌酐、膽固醇及甘油三酯均相對TBC染毒組有極大的改善。表4血液生化指標檢測結果a注:﹡與對照組比較,p<0.05;﹡﹡與對照組比較,p<0.01.②表5給出了堿性磷酸酶(ALP),丙氨酸轉氨酶(ALT),天冬氨酸轉氨酶(AST),總蛋白(TP),白蛋白(ALB),總膽紅素(TBIL),總膽汁酸(TBA),總膽固醇(TCHO),三酰甘油(TG),葡萄糖(GLU)的檢測結果,從表5可以看出表5血液生化指標檢測結果b注:﹡與對照組比較,p<0.05.(三)三十天的喂養試驗結束后,動物各臟器稱重,計算臟體比值,結果見表6,從表6可以看出,雄性大鼠和雌性大鼠染毒組的50mg/kg·bw和100mg/kg·bwPSPP干預組與對照組的肝/體比值比較TBC染毒組有明顯的改善,存在統計學差異,且有劑量效應關系。表6染毒四周后肝臟/體比結果注:﹡與對照組比較,p<0.05.(四)三十天的喂養試驗結束后,取部分肝臟備做病理組織學檢測,得到如圖8~11所示的肝組織病理學切片(x400)照片,從圖8~11可以看出,結果發現染毒組和低劑量PSPP干預組相對高劑量PSPP干預組和對照組在病理組織學檢查時發現肝臟出現空泡和炎細胞浸潤,表明PSPP干預組對肝臟有改善和保護作用。(五)三十天的喂養試驗結束后,在低溫條件下制備肝勻漿檢測超氧化物歧化酶(SOD),過氧化氫酶(CAT),谷胱甘肽過氧化物酶(GSHPx),丙二醛(MDA)等指標,結果見表7,從表7可以看出,比較上述各項指標兩組PSPP干預組相比染毒組均有很大改善。表7抗氧化指標檢測結果注:*與對照組比較,p<0.05;**與對照組比較,p<0.01.甘薯多糖干預TBC致肝損傷的體內HepG2細胞凋亡抑制試驗過程如下:以HepG2細胞為靶細胞,以TBC誘導HepG2細胞凋亡的劑量處理細胞后,再用不同劑量和不同處理時間來觀察PSPP對TBC致HepG2細胞凋亡的保護作用,闡明其具有劑量效應及時間效應關系。得到如表8所示的PSPP干預對HepG-2細胞的凋亡率結果,從表8可以看出,PSPP干預組的凋亡率比較TBC染毒組,其凋亡率有明顯的降低。表8流式細胞儀檢測PSPP干預對HepG-2細胞的凋亡率組別細胞數濃度凋亡率(%)對照組1×106--0.97±0.29PSPP干預組1×1065mmol·L-111.83±0.55**PSPP干預組1×1062.5mmol·L-115.98±1.52**TBC染毒組1×10610mmol·L-132.28±1.21**注:﹡與對照組比較,p<0.05,﹡﹡與對照組比。當前第1頁1 2 3
相關技術
網友詢問留言
已有0條留言
- 還沒有人留言評論。精彩留言會獲得點贊!
1