一種熱原子層沉積技術生長GeTe合金薄膜的方法與流程
本發明涉及一種熱原子層沉積技術生長gete合金薄膜的方法,屬于半導體制備技術領域。
背景技術:
隨著科技的進步,半導體技術得到快速的發展,其中微電子器件中的存儲器的類型及制作工藝技術隨之發生了變革。相變存儲器(pcram)融合了動態隨機存儲器(dram)的高容量、低成本及靜態隨機存儲器(sram)的高速度、低電壓、低功耗等優勢,成為國際上公認的下一代的信息存儲器。相變材料的核心組成為硫屬合金薄膜,其中ge-sb-te三元體系中的gesbte225薄膜以最為優良的綜合性能倍受青睞。尤其是gete合金薄膜的制備是半導體存儲器行業中的關鍵技術。
在已公開的專利中,相較于pvd、cvd技術,ald技術具有明顯優勢,制得的相變材料薄膜的保型性較好,但是仍然存在沉積過程中源之間化學反應活性低、雜元素污染薄膜及工藝條件苛刻等問題。
ald原理依賴于交替的脈沖,將前驅體蒸汽分別傳送到反應室中基質材料的表面,隨后發生化學吸附和界面反應的過程。在前驅體蒸汽以脈沖的形式進入沉積室后,用惰性氣體對沉積室進行清洗凈化,以此循環,進而根據循環次數控制所沉積薄膜的厚度。成功的ald過程嚴重依賴于合適的前驅體,ald前驅體可以是氣體、液體或揮發性固體材料,其中液態前驅體為最佳選擇。首先,氣態前驅體不易控制,在高溫條件下存在一定危險;然后,在ald成膜過程中需要短時間內維持恒定的前驅體脈沖,而固態前驅體的揮發依賴于粒徑大小,會導致前驅體脈沖不規律地變化,另外,固體前驅體在揮發過程中其表面會有雜質集中的傾向,會阻礙揮發。相比之下,液態前驅體是一個表面成分不斷更新揮發的過程,能夠維持恒定的表面積,有恒定持續的蒸氣壓,能減少前驅體的浪費,也能夠更好的控制操作過程。
更具體地,理想的ald的前驅體應該具備如下性質:
(1)具有較好的揮發性,即具有較低的揮發溫度,在稍微加熱或不加熱的情況下,就能獲得足夠高的蒸汽壓,將前驅體有效的傳輸到反應室內并達到沉積薄膜所需要的蒸汽壓;(2)具有足夠的熱穩定性,即保證將前驅體傳輸到反應室的過程中及在基質表面不發生分解;(3)具有較高的化學反應活性,即前驅體必須在化學吸附或與材料表面基團快速有效反應進而沉積薄膜;(4)具有較高的化學純度,避免雜元素的摻雜影響薄膜的性能及器件壽命;(5)利于儲存運輸,在正常條件下能盡可能長時間的儲存且不發生分解,并在運輸過程中不會發生易燃易爆的現象;(6)無毒無害,危險系數低;(7)廉價易得。
雖然,在cvd技術的基礎上進化出的ald技術可以通過合適前驅體的選擇,在基質板上發生飽和的界面化學反應,實現薄膜自限制生長的特性。但是目前,ald沉積ge及其合金薄膜仍受到ge前驅體缺乏的限制。
由于烷基/氨基ge配合物的化學穩定性很高,化學反應活性低,與其他前驅體之間缺乏靈活的配體交換反應,所以多被用于cvd方法熱分解沉積ge/ge合金薄膜。目前,僅有較少烷基/氨基ge配合物被用作ald前驅體沉積gst的文獻報道。
2007年,choibjetal分別ge(ibu)4、sb(ipr)3、te(ipr)2作為ge、sb、te的前驅體,成功制備gst薄膜,但僅有ge膜的生長過程呈現出飽和生長的特性。2007年,j.leeetal分別選用ge(n(tms)2)4/ge(nme2)4、sb(nme2)3、te(c3h7)2作為ge、sb、te的前驅體成功制備gst薄膜,但是存在高濃度的si、c、n的殘留。另外ge(nme2)4的合成過程中涉及易燃且有腐蝕性二甲氨基鋰;ge[n(tms)2]4作為前驅體成膜時生長速率緩慢等,也都阻礙了此類ge前驅體的進一步應用。2015年,songetal.用ge(nme2)4、sb(nme2)3、te(tbu)2作為ge、sb、te的的前驅體,沉積得到gst225和gst181晶態膜,依然存在高濃度的c、n的殘留。以上雖然都采用了ald技術,仍然是利用了前驅體的分解成膜,不能滿足pcram對薄膜性能的要求。
直到烷硅基碲作為前驅體被應用到ald中,才使得ald的自限制反應沉積gt/gst薄膜的特性得以顯現。這主要是由于te在烷基碲中為+2價,正價態的te化合物難以與正價態的ge配合物進行反應,所以烷基碲多被用于熱分解沉積合金薄膜。而te在烷硅基碲中顯-2價,較容易與正價態的ge配合物反應。為實現ge、te前驅體之間化學反應,proeetal.利用鍺的氯化物為前驅體,引入si-cl路易斯酸堿對,實現ge、te前驅體之間靈活的配體交換。自此,激發了研究者們對ald沉積gt/gst薄膜的熱情。
2009年,proeetal.分別使用gecl2(c4h8o2)、sbcl3、(et3si)2te作為ald的ge、sb、te前驅體。在90℃下,通過配體交換反應(反應方程式見圖1的式a)實現每一個沉積過程的飽和界面反應過程,成功制備具有優秀保型性的gst225薄膜。
2012年,eometal.分別用ge(ome)4、sb(oet)3、(tms)2te為ald的ge、sb、te前驅體。在70℃下,ald過程沉積出gst227膜(反應方程式見圖1的式b),但是不能滿足pcm對gst三元合金化學比例的要求。這是因為ge原子在ge(ome)4為+4價,而te原子在(tms)2te中為-2價,當這兩種化合物發生配體交換反應時會生成gete2,所以用+4價的ge化合物作為ald的前驅體不能直接得到gst225薄膜。
為了解決ald沉積gst薄膜的化學比例的問題,2016年,eometal.選用ge(n(tms)2)2、(tms)2te為ald前驅體,并以meoh為還原劑。在70-120℃的操作窗口下沉積得到gete薄膜(反應方程式見圖1的式c)
上述的方法,有的雖然是ald方法,但沒有ald之實;目前僅有proeetal、eometal.實現了純粹的ald沉積gt/gst薄膜過程。他們選用的ge前驅體不同,但同樣采用路易斯酸堿反應的理論基礎,目的在于形成si-cl、si-o路易斯酸堿對。這就不可避免的造成薄膜中o、cl雜元素的摻雜,影響薄膜的性能及器件壽命。另外pore等所使用的gecl2(c4h8o2)前驅體本身熱穩定性差,高溫下易在薄膜表面形成顆粒,而且生成的氯化物會對設備造成腐蝕。eom等使用的ge(ome)4前驅體難以得到gst225的化學比例的薄膜;ge(n(tms)2)2的熱穩定性差等,都限制了這些ge前驅體的應用。
技術實現要素:
為了克服上述現有技術存在的缺點,本發明提供了一種適用于ald沉積技術制備gete合金薄膜的ge前驅體化合物以及利用該化合物進行熱原子層沉積技術生長gete合金薄膜的方法。本發明中的方法使用了合適的源前驅體組合,極大的提高了源之間的化學反應活性、有效的避免了雜元素的摻雜,制得的薄膜的化學組份單一,另外,在溫和的工藝條件下,在納米級的半導體器件上沉積形成保型性優良的gete沉積層。
本發明的熱原子沉積技術生長gete合金薄膜的方法,包括以下步驟:a)將襯底置于反應腔中,在真空條件下,以脈沖形式向反應腔中通入氣相ge源進行沉積,得到沉積有ge源的襯底,所述ge源包括具有式i所示結構的化合物;b)將氣相還原劑以脈沖形式通入反應腔,對沉積在襯底上的ge源進行還原得到中間物質;c)以脈沖的形式向反應腔中通入氣相te源,與沉積在襯底上的中間物質進行反應得到gete合金薄膜的襯底;其中所述ge源為具有式i所示結構的化合物;
其中r1表示氫原子、c1~c6烷基、c2~c5鏈烯,r2,r5表示氫原子、c1~c6烷基、c2~c5鏈烯基、c6~c10芳基或-si(r6)3,r3,r4表示-si(r6)3,其中r6為c1~c6烷基;r1,r2,r5相同或相異,r3,r4相同或相異。
在本發明的一種實施方式中,所述式i的化合物中,r1表示甲基或nbu,r3、r4表示-si(r6)3且r6為甲基,r2、r5表示仲丁基sbu或者異丙基ipr,且r2、r5相同或相異。
在本發明的一種實施方式中,所述te源為具有式ⅱ所示結構的化合物:
其中r7,r8為c1~c6烷基,r7,r8相同或相異。
在一種實施方式中,所述步驟a)中以脈沖形式向反應腔中通入氣相ge源的單個脈沖的持續時間為0.05~20s。
在一種實施方式中,所述步驟a)中兩個脈沖之間的間隔時間為0.5~30s。
在一種實施方式中,所述步驟a)中的沉積的溫度為60~400℃。
在一種實施方式中,所述氣相ge源在載氣存在條件下以脈沖形式通入;載氣的流量為10~200sccm。
在一種實施方式中,所述步驟b)中氣相還原劑包括h2、nh3、b2h6、單烷基硼烷、氨基硼烷、醇類、肼類、烷基鋁、氨基鋁烷類和烷基鋅中的一種或幾種。
在一種實施方式中,所述步驟b)中將氣相還原劑以脈沖形式通入反應腔的單個脈沖的持續時間為0.01~20s。
在一種實施方式中,所述步驟b)中兩個脈沖之間的間隔時間為0.5~30s。
在一種實施方式中,所述步驟b)中氣相還原劑在載氣存在的條件下以氣相脈沖形式通入;所述載氣的流量為10~200sccm。
在一種實施方式中.,所述步驟c)中以脈沖形式向反應腔中通入氣相te源的單個脈沖的持續時間為0.05~20s。
在一種實施方式中,所述步驟c)中兩個脈沖之間的間隔時間為0.5~30s。
在一種實施方式中,所述步驟c)中的沉積的溫度為60~400℃。
在一種實施方式中,所述氣相te源在載氣存在條件下以脈沖形式通入;所述載氣的流量為10~200sccm。
在一種實施方式中,所述半導體襯底包括硅、氧化硅、氮化硅、tan和藍寶石中的一種或幾種。
本發明的第二個目的是提供一種ge源化合物,其結構是如式i所示:
其中r1表示氫原子、c1~c6烷基、c2~c5鏈烯;r2,r5表示氫原子、c1~c6烷基、c2~c5鏈烯基、c6~c10芳基或-si(r6)3;r3,r4表示-si(r6)3,其中r6為c1~c6烷基;r1,r2,r5相同或相異;r3,r4相同或相異。
在本發明的一種實施方式中,所述式i的化合物中,r1表示甲基或nbu,r3、r4表示-si(r6)3且r6為甲基,r2、r5表示仲丁基sbu或者異丙基ipr,且r2、r5相同或相異。
本發明的有益效果:
本發明的具有式i中的ge源較目前應用于ald的源如二氯化鍺二氧六環、四(烷氧基)鍺和二(二三甲基硅基氨基)鍺具有如下優勢:
(1)揮發性與二氯化鍺二氧六環相仿,但熱分解溫度遠高于二氯化鍺二氧六環;本申請中的ge源的熱分解溫度大于300℃,且殘余質量幾乎為零,在提純過程中發現可以在150℃的高溫下,1d內不會發生任何熱分解,能夠適用于較高溫度的ald過程;
(2)二氯化鍺二氧六環中含有cl元素,在高溫條件下生成的氯化物不僅污染薄膜且腐蝕儀器,另外還含有o元素也會造成薄膜中o元素的摻雜而導致影響薄膜的性能及器件壽命;而本申請中的ge源前驅體分子中無o、cl等對薄膜性能造成不良影響的雜元素,且該ge源前驅體該對空氣濕度敏感程度低,易于存儲及運輸。
(3)室溫下,二氯化鍺二氧六環為固態,高溫下易分解,而本申請中的ge源為液態,且熱穩定性良好,易于獲得滿足電子元器件對高純源的要求,適用于大規模生產;
(4)本申請中的ge源前驅體能夠被多種液態還原劑還原生成中間物質,然后與te源前驅體反應制得gete合金薄膜,相對于目前常用的h2或nh3更易于操作、更安全。
進一步的,具有式i中化合物較其他結構類似的ge配合物,如(n,n’-二異丙基-二三甲基硅基胍基)(六甲基二硅氮基)ge具有如下優勢:
(1)胍基鍺的熱穩定性差與脒基鍺,且合成麻煩,成本更高;
(2)(n,n’-二異丙基-二三甲基硅基胍基)(六甲基二硅氮基)ge為固態;而本申請的式i化合物為液態,更易在保證純度的前提下工業化生產;液態前驅體為ald前驅體的最佳選擇。在ald成膜過程中需要短時間內維持恒定的前驅體脈沖,而固態前驅體的揮發依賴于粒徑大小,會導致前驅體脈沖不規律地變化;另外,固體前驅體在揮發過程中其表面會有雜質集中的傾向,會阻礙揮發。相比之下,液態前驅體是一個表面成分不斷更新揮發的過程,能夠維持恒定的表面積,有恒定持續的蒸氣壓,能減少前驅體的浪費,也能夠更好的控制操作過程。
本發明的方法具有如下優點:
(1)選用了合適的前驅體組合,提高了源之間的化學反應活性、有效的避免了雜元素的摻雜,制得的薄膜的化學組份單一;所制備得到的gete合金薄膜為高電阻率的非晶態薄膜;gete合金薄膜的電阻在1.4~4*105ω·cm,薄膜的均方根粗糙度在0.7nm。
(2)對多種襯底如硅、氧化硅、氮化硅、tan、藍寶石等均表現出兼容性。
(3)選用的源都是易揮發的液態源,且都具有良好的熱穩定性,有效地降低了沉積成膜的工藝難度,因此能夠適用于較高溫度的熱原子層(t-ald)沉積過程,制得高純且保型性較好的gete沉積層。
(4)在溫和的工藝條件下,使用t-ald技術在納米級的半導體器件上沉積形成保型性優良的gete沉積層。
(5)本發明提供的熱原子沉積技術生長gete合金薄膜的方法有效地克服現有的技術缺點,提高了源之間的化學反應活性,保證了薄膜中化學組分的純凈單一性,有效地降低了沉積工藝的難度,廣泛地應用于半導體存儲和微電子技術領域。
附圖說明
圖1為反應方程式;
圖2為(n,n’-二仲丁基-甲基脒基)(六甲基二硅氮基)ge(ⅱ)的h-nmr;
圖3為(n,n’-二仲丁基-甲基脒基)(六甲基二硅氮基)ge(ⅱ)與二氯化鍺二氧六環的熱分解圖;
圖4為實施例2中的gete合金薄膜的eds圖片;
圖5為實施例2中的gete合金薄膜的sem圖片;
圖6為實施例2中的gete合金薄膜的afm圖片;
圖7為(n,n’-二異丙基-正丁基脒基)(六甲基二硅氮基)ge(ⅱ)的h-nmr;
圖8為式v化合物的h-nmr。
具體實施方案
為了更清楚地說明本發明實施例或現有技術中的技術方案,下面將對實施例或現有技術描述中所需要使用的附圖作簡單地介紹,顯而易見地,下面描述中的附圖僅僅是本發明的實施例,對于本領域普通技術人員來講,在不付出創造性勞動的前提下,還可以根據提供的附圖獲得其他的附圖。
本發明提供了一種熱原子沉積技術生長gete合金薄膜的方法,包括以下步驟:
a)將襯底置于反應腔中,在真空條件下,以脈沖形式向反應腔中通入氣相ge源進行沉積,得到沉積有ge源的襯底,所述ge源包括具有式i所示結構的化合物;
其中r1表示氫原子、c1~c6烷基、c2~c5鏈烯;r2,r5表示氫原子、c1~c6烷基、c2~c5鏈烯基、c6~c10芳基或-si(r6)3;r3,r4表示-si(r6)3,其中r6為c1~c6烷基;r1,r2,r5相同或相異,r3,r4相同或相異。
b)將氣相還原劑以脈沖形式通入反應腔,對沉積在襯底上的ge源進行還原得到中間物質;
c)以脈沖的形式向反應腔中通入氣相te源,與沉積在襯底上的ge的中間物質進行反應得到gete合金薄膜,所述te源包括具有式ⅱ所示結構的化合物。
其中r7,r8為c1~c6烷基;r7,r8相同或相異。本發明將襯底置于反應腔中,在真空條件下,以脈沖形式向反應腔中通入氣相ge源進行沉積,得到沉積有ge源的襯底,本發明優選先將所述需要沉積含gete合金薄膜的襯底進行清洗,得到預處理的襯底。在本發明中,優選使用工業界標準清洗,如,使用spm(h2so4/h2o2)溶液去除襯底表面的有機沾污,使用apm(nh4oh/h2o2)溶液去除襯底表面的顆粒沾污,采用稀釋的hf溶液漂洗去除襯底表面的自然氧化層。在實際應用中,不限于此種清洗方法,也可視實際應用使用其它清洗方法,如丙酮-異丙醇清洗等。
得到預處理的襯底后,本發明優選將預處理的襯底放入原子層沉積設備的傳片腔并抽真空,實現沉積所需的真空環境,達到要求的真空度后,再傳入反應腔,以避免空氣中的水氧擴散至反應腔影響金屬膜的生長。為了進一步的保證原子層沉積設備中各管路及腔體內無水氧殘留,在放置襯底前,本發明優選對原子層沉積設備的管路及反應腔體進行抽空或預長膜處理。
在本發明中,襯底優選包括硅、氧化硅、氮化硅、tan和藍寶石中的一種或幾種;所述氣相ge源包括具有式i所示結構的化合物,該化合物化學式包括(n,n’-二仲丁基-甲基脒基)(六甲基二硅氮基)ge(ⅱ)、(n,n’-二異丙基-正丁基脒基)(六甲基二硅氮基)ge(ⅱ)、(n-仲丁基-n’-正丙基-甲基脒基)(六甲基二硅氮基)ge(ⅱ)、(n-仲丁基-n’-異丙基-甲基脒基)(六甲基二硅氮基)ge(ⅱ)、(n-仲丁基-n’-正丁基-甲基脒基)(六甲基二硅氮基)ge(ⅱ)和(n-仲丁基-n’-叔丁基-甲基脒基)(六甲基二硅氮基)ge(ⅱ)中的一種或幾種。所述氣相te源包括具有式ⅱ所示結構的化合物,該化合物化學式為(et3si)2te,本發明對具有式ⅱ所示結構的te源沒有特殊的限制,可以按照參考文獻ecstransactions,2009,25(8):609-616.進行合成。
本發明優選對ge源進行加熱,使之氣化,得到氣相ge源,所述對ge源加熱的溫度優選為25~200℃,更優選為50~180℃,具體的,可以是50℃、100℃或150℃。
其中r1表示氫原子、c1~c6烷基、c2~c5鏈烯;r2,r5表示氫原子、c1~c6烷基、c2~c5鏈烯基、c6~c10芳基或-si(r6)3;r3,r4表示-si(r6)3,其中r6為c1~c6烷基;r1,r2,r5相同或相異;r3,r4相同或相異。
在本發明中,所述氣相ge源的單個脈沖的持續時間優選為0.05~20s,更優選為1~18s,最優選為3~15s,具體的,在本發明的實施例中,可以是1s、5s、8s、12s或16s;所述氣相ge源兩個脈沖之間的間隔時間優選為0.5~30s,更優選為1~25s,最優選為5~20s,具體的,在本發明的實施例中,可以是5s、10s、15s、20s或25s;所述沉積的溫度優選為50~300℃,更優選為70~250℃,最優選為90~200℃,具體的,在本發明的實施例中,可以是70℃、90℃、125℃、200℃或250℃;所述氣相ge源的的載氣優選為高純氮氣或高純氬氣,所述載氣的流量優選為10~200sccm,更優選為20~160sccm,最優選為60~120sccm,具體的,可以是20sccm、90sccm、120sccm、160sccm或60sccm。
完成一次ge源的沉積后,本發明優選采用高純氮氣或高純氬氣對反應腔體進行吹掃清洗,清洗的時間優選為5~50s,更優選為10~45s,最優選為15~40s。
然后,本發明將氣相還原劑以氣相脈沖形式通入反應腔內,對沉積在襯底上的ge源進行還原,得到沉積有含ge中間物質的襯底,在本發明中,所述還原劑優選包括h2、nh3、b2h6、單烷基硼烷、氨基硼烷、醇類、肼類、烷基鋁、氨基鋁烷類和烷基鋅中的一種或幾種,更優選包括h2、nh3、b2h6、單烷基硼烷(r1bh2或r1r2bh)、氨基硼烷(r1r2hn·bh3或r1r2r3n·bh3)、醇類(r1oh)、肼類(r1nhnh2或n2h4)、烷基鋁(alr1r2r3)、氨基鋁烷類(r1r2r3n·alh3)和烷基鋅(znr1r2)中的一種或幾種其中r1,r2,r3為c1~c10烴基,且三者可以相同也可以不同,不同物質中的可以相同也可以不同,如r1oh和r1nhnh2中的r1可以相同也可以不同。具體的,在本發明的實施例中,還原劑可采用n2h4、me2nh·bh3、ch3oh、alme3或znet2。本發明優選將所述還原劑加熱,使之氣化,形成氣態的還原劑。所述加熱還原劑的溫度優選為25~150℃,更優選為40~140℃,具體的,在本發明的實施例中,可以是60℃、90℃、25℃或85℃。
在本發明中,所述通入還原劑的單個脈沖的持續時間優選為0.01~20s,更優選為1~15s,更優選為5~10s,具體的,在本發明的實施例中,可以是10s、1s、20s、15s或5s;所述通入還原劑兩個脈沖之間的間隔時間優選為0.5~30s,更優選為1~25s,最優選為5~20s,具體的,在本發明的實施例中,可以是15s、5s、10s、25s或20s。所述氣相還原劑的載氣優選為高純氮氣或高純氬氣,所述載氣的流量優選為10~200sccm,更優選為20~160sccm,最優選為60~120sccm。
完成一次還原后,本發明優選采用高純氮氣或高純氬氣對反應腔體進行吹掃清洗,所述清洗的時間優選為5~50s,更優選為10~45s,最優選為15~40s。
在本發明中,所述氣相te源的單個脈沖的持續時間優選為0.05~20s,更優選為1~18s,最優選為3~15s,具體的,在本發明的實施例中,可以是1s、5s、8s、12s或16s;所述氣相te源兩個脈沖之間的間隔時間優選為0.5~30s,更優選為1~25s,最優選為5~20s,具體的,在本發明的實施例中,可以是5s、10s、15s、20s或25s;所述沉積的溫度優選為50~300℃,更優選為70~250℃,最優選為90~200℃,具體的,在本發明的實施例中,可以是70℃、90℃、125℃、200℃或250℃;所述氣相源的的載氣優選為高純氮氣或高純氬氣,所述載氣的流量優選為10~200sccm,更優選為20~160sccm,最優選為60~120sccm,具體的,可以是20sccm、90sccm、120sccm、160sccm或60sccm。
完成一次te源的沉積后,本發明優選采用高純氮氣或高純氬氣對反應腔體進行吹掃清洗,清洗的時間優選為5~50s,更優選為10~45s,最優選為15~40s。
本發明優選重復上述氣相ge源沉積-吹掃清洗-氣相還原劑還原-吹掃清洗-氣相te源沉積-吹掃起清洗這一過程,重復循環的次數視實際需求而定,在本發明中,所述循環的次數優選為300~4500次,更優選為1000~3000次,具體的,在本發明的實施例中,可以是300次、1000次、1500次、3000次或4500次。
為了進一步說明本發明,以下結合實施例對本發明提供的一種熱原子層沉積技術生長gete合金薄膜的方法進行詳細描述,但不能將其理解為對本發明保護范圍的限定。
實施例1:(n,n’-二仲丁基-甲基脒基)(六甲基二硅氮基)ge(ⅱ)的制備
式iii所示的(n,n’-二仲丁基-甲基脒基)(六甲基二硅氮基)ge(ⅱ)的制備方法如下:
100mlschlenk瓶抽真空充氬氣置換三次,氬氣保護下加入n,n’-二仲丁基-甲基脒配體(0.510g,3mmol)、重蒸除水的乙醚(15ml),體系為無色溶液。在-78℃下,逐滴滴加正丁基鋰(1.2ml,3mmol)。緩慢恢復室溫,并繼續攪拌3h,體系為淡黃色溶液。
另取100mlschlenk瓶抽真空充氬氣置換三次,氬氣保護下加入二氯化鍺二氧六環(0.693g,3mmol)、重蒸除水的乙醚(10ml)。在氬氣氣體保護、-78℃下,將鋰鹽的乙醚溶液逐滴滴加至二氯化鍺二氧六環的乙醚液中。緩慢恢復室溫后并繼續攪拌12h。
另取100mlschlenk瓶抽真空充氬氣置換三次,在氬氣氣體保護下,加入六甲基二硅氮烷(0.484g,3mmol)、重蒸除水的乙醚(15ml),體系為無色溶液。在-78℃下,逐滴滴加正丁基鋰(1.2ml,3mmol)。緩慢恢復室溫后繼續攪拌3h,體系為無色溶液。
在氬氣氣體保護、-78℃下,將上述鋰鹽的乙醚溶液緩慢滴加至上述單取代的鍺配合物的乙醚液中。緩慢恢復室溫后并繼續攪拌12h。真空條件除去體系中易揮發組份,并用15ml*2重蒸除水的正己烷提取產物。真空條件除去提取液中易揮發組份,得到淡黃色油狀粗產物。提純:真空條件、油浴溫度120℃下,用微量減壓蒸餾裝置提純得到無色油狀液態產物0.399g(產率約51%)。
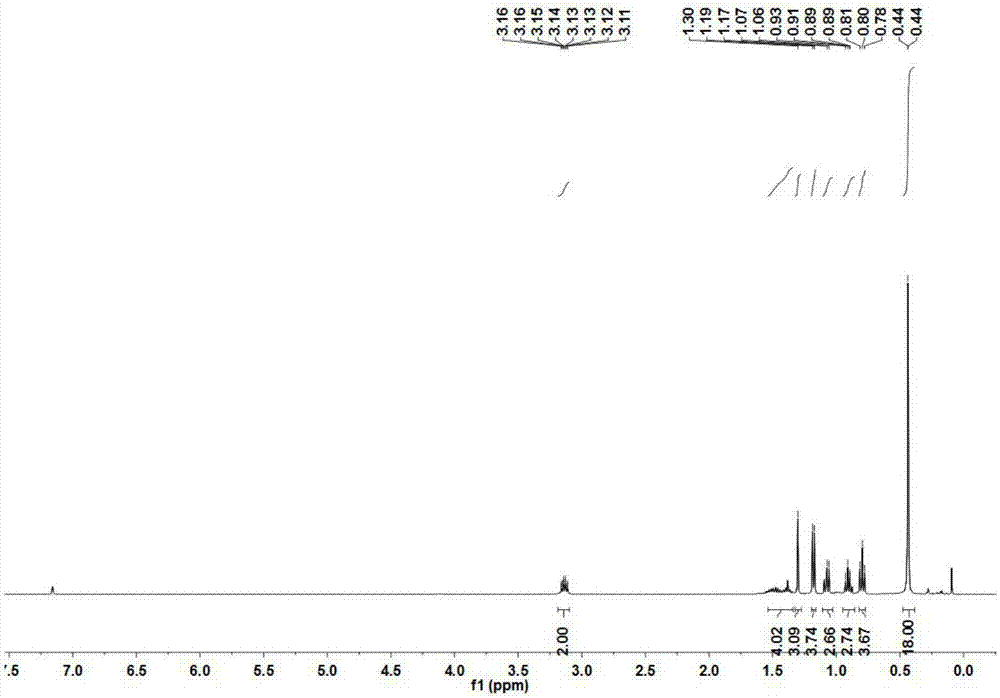
其中:1hnmr(400mhz,c6d6)δ3.14(ddd,j=12.9hz,2h),1.44–1.33(m,4h),1.30(s,3h),1.18(d,j=6.4hz,3h),1.06(d,j=6.4hz,3h),0.91(dd,j=10.5hz,3h),0.80(t,j=7.4hz,3h),0.44(d,j=0.8hz,18h),見附圖2。
在結構確認后,進行了熱化學性質測試,如圖3所示為本實施例中的ge源與二氯化鍺二氧六環的熱分解圖。由圖3可以看出,二氯化鍺二氧六環在170℃開始分解,殘余質量較高,在成膜過程中分解形成顆粒物摻雜在薄膜中;而本實施例中的ge源熱分解溫度遠高于二氯化鍺二氧六環。本實施例中的ge源,初始揮發溫度為165℃,t50(失重50%時的溫度)為190℃,終止揮發溫度為210℃,揮發過程為單階揮發曲線、無多余拐點,且最終殘余質量僅為1.2%,說明(n,n’-二仲丁基-甲基脒基)(六甲基二硅氮基)ge(ⅱ)具有良好的揮發性;本實施例中的ge源,熱分解溫度大于300℃(300℃下ge源未發生分解),且殘余質量幾乎為零;所以該ge源化合物熱穩定性好,適用于ald前驅體。
實施例2:基于原子層沉積的gete合金薄膜的制備
一種以(n,n’-二仲丁基-甲基脒基)(六甲基二硅氮基)ge(ⅱ)為ge源,見具有式iii,以n2h4為還原劑,以(et3si)2te為te源制備gete合金薄膜原子層沉積方法,包括以下過程:
1)以si為襯底,沉積溫度為150℃,ge源(n,n’-二仲丁基-甲基脒基)(六甲基二硅氮基)ge(ⅱ)的加熱溫度為50℃,使之氣化,以高純氮氣為載氣,通入氣相ge源(n,n’-二仲丁基-甲基脒基)(六甲基二硅氮基)ge(ⅱ),載氣流量為20sccm。脈沖時間為12s,等待時間為10s;
2)完成一個ge源脈沖后使用高純氮氣進行清洗,清洗時間為25s;
3)還原劑n2h4加熱溫度為40℃,使之氣化,以高純氮氣為載氣,載氣流量為60sccm,以脈沖的形式通入n2h4脈沖時間為5s,等待時間為15s。
4)完成一個還原劑脈沖后采用高純氮氣進行清洗,清洗時間為15s。
5)te源(et3si)2te的加熱溫度為45℃,使之氣化,以高純氮氣為載氣,通入氣相te源(et3si)2te,載氣流量為20sccm。脈沖時間為12s,等待時間為10s;
6)完成一個te源脈沖后使用高純氮氣進行清洗,清洗時間為45s;
將上述1)~6)步驟重復循環300次,所得gete合金薄膜厚度為10nm,采用四探針法測試電阻率為4*105ω·cm。
本發明對本實施例得到的gete合金薄膜進行x射線能譜儀測試,結果如圖4所示,圖4為本發明實施例2中的薄膜的圖片,由圖4可以看出,本實施例得到的gete合金薄膜薄膜具有純凈的化學組份,無其他雜元素的摻雜,其中si元素的出現是薄膜層較薄致使電子束轟擊到了硅片表面而檢測到元素si。對本實施例得到的gete合金薄膜進行掃描電鏡測試,結果如圖5所示,圖5為本發明實施例2中的薄膜的圖片,由圖5可以看出,本實施例得到的gete合金薄膜薄膜具有良好的臺階覆蓋性,無堆積成島現象,由此表明該方法制備的gete合金薄膜具有良好的保型性。對本實施例得到的gete合金薄膜進行原子力學顯微鏡測試,結果如圖6所示,由圖6可以看出,本實施例得到的gete合金薄膜的均方根粗糙度在0.7nm,表明該方法制備gete合金的薄膜的表面形態較好。
實施例3
一種以(n,n’-二異丙基-正丁基脒基)(六甲基二硅氮基)ge(ⅱ)為ge源,見具有式iv,以n2h4為還原劑,以(et3si)2te為te源制備gete合金薄膜原子層沉積方法,包括以下過程:
1)以sio2為襯底,沉積溫度為100℃,ge源(n,n’-二異丙基-正丁基脒基)(六甲基二硅氮基)ge(ⅱ)的加熱溫度為70℃,使之氣化,以高純氬氣為載氣,通入氣相ge源(n,n’-二異丙基-正丁基脒基)(六甲基二硅氮基)ge(ⅱ),載氣流量為50sccm。脈沖時間為5s,等待時間為20s;
2)完成一個ge源脈沖后使用高純氬氣進行清洗,清洗時間為45s;
3)還原劑meoh加熱溫度為70℃,使之氣化,以高純氬氣為載氣,載氣流量為10sccm,以脈沖的形式通入meoh脈沖時間為15s,等待時間為5s。
4)完成一個還原劑脈沖后采用高純氬氣進行清洗,清洗時間為35s。
5)te源(et3si)2te的加熱溫度為60℃,使之氣化,以高純氬氣為載氣,通入氣相te源(et3si)2te,載氣流量為40sccm。脈沖時間為20s,等待時間為25s;
6)完成一個te源脈沖后使用高純氬氣進行清洗,清洗時間為45s;
將上述1)~6)步驟重復循環1000次,所得gete合金薄膜厚度為27nm,采用四探針法測試電阻率為1.4*105ω·cm。得到的gete合金薄膜薄膜具有純凈的化學組份,無其他雜元素的摻雜,具有良好的保型性。
實施例4
一種以(n-仲丁基-n’-異丙基-甲基脒基)(六甲基二硅氮基)ge(ⅱ)為ge源,見具有式v,以alme3為還原劑,以(et3si)2te為te源制備gete合金薄膜原子層沉積方法,包括以下過程:
1)以氮化硅為襯底,沉積溫度為80℃,ge源(n-仲丁基-n’-異丙基-甲基脒基)(六甲基二硅氮基)ge(ⅱ)的加熱溫度為90℃,使之氣化,以高純氬氣為載氣,通入氣相ge源(n-仲丁基-n’-異丙基-甲基脒基)(六甲基二硅氮基)ge(ⅱ),載氣流量為100sccm。脈沖時間為8s,等待時間為5s;
2)完成一個ge源脈沖后使用高純氬氣進行清洗,清洗時間為15s;
3)還原劑alme3加熱溫度為50℃,使之氣化,以高純氬氣為載氣,載氣流量為120sccm,以脈沖的形式通入alme3脈沖時間為1s,等待時間為25s。
4)完成一個還原劑脈沖后采用高純氬氣進行清洗,清洗時間為45s。
5)te源(et3si)2te的加熱溫度為70℃,使之氣化,以高純氬氣為載氣,通入氣相te源(et3si)2te,載氣流量為45sccm。脈沖時間為25s,等待時間為45s;
6)完成一個te源脈沖后使用高純氬氣進行清洗,清洗時間為25s;
將上述1)~6)步驟重復循環1500次,所得gete合金薄膜厚度為16nm,采用四探針法測試電阻率為0.9*105ω·cm。得到的gete合金薄膜薄膜具有純凈的化學組份,無其他雜元素的摻雜,具有良好的保型性。
實施例5:式iv化合物的制備及結構確認
(n,n’-二異丙基-正丁基脒基)(六甲基二硅氮基)ge(ⅱ)的制備:
取100mlschlenk瓶抽真空充氬氣置換三次,氬氣保護下,加入n,n'-二異丙基碳二亞胺(4.086g,32.3mmol)、重蒸除水的正己烷(50ml),體系為無色透明液體。然后在-78℃下逐滴滴加正丁基鋰(14ml,35mmol),無沉淀生成,為淡黃色澄清透明溶液,緩慢升至室溫反應直至有白色沉淀析出。通過無水無氧過濾洗滌裝置在氬氣保護下將反應體系過濾,20ml*2重蒸除水的正己烷洗滌白色固體,真空干燥后得到白色固體(5.547g,產率約90%,熔點:128~131℃),放置在手套箱中保存待取用。
手套箱中取n,n’-二異丙基-正丁基脒基鋰鹽(0.369g,1.5mmol)于100mlschlenk瓶中。氬氣保護下,加入重蒸除水的乙醚(15ml)并攪拌20min,體系為無色溶液。在-78℃下,將鋰鹽的乙醚溶液逐滴滴加至二氯化鍺二氧六環(0.346g,1.5mmol)的乙醚(10ml)懸濁液中。緩慢恢復室溫后并繼續攪拌12h。
另取100mlschlenk瓶抽真空充氬氣置換三次,在氬氣氣體保護下,加入六甲基二硅氮烷(0.242g,,1.5mmol)、重蒸除水的乙醚(15ml),體系為無色溶液。在-78℃下,逐滴滴加正丁基鋰(0.6ml,1.5mmol)。緩慢恢復室溫后繼續攪拌3h,體系為無色溶液。
在氬氣氣體保護、-78℃下,將上述鋰鹽的乙醚溶液逐滴滴加至上述單取代的鍺配合物的乙醚液中。緩慢恢復室溫后并繼續攪拌12h。真空條件除去體系中易揮發組份,并用15ml*2重蒸除水的正己烷提取產物。真空條件除去提取液中易揮發組份,得到淡黃色油狀粗產物。提純:真空條件、油浴溫度120℃下,用微量減壓蒸餾裝置提純得到無色油狀液態產物0.399g(產率約51%)。
其中,1hnmr(400mhz,c6d6)δ3.49(dt,j=12.7,6.3hz,2h),1.90–1.74(m,2h),1.36–1.26(m,4h),1.29–1.13(m,12h),0.77(t,j=7.3hz,3h),0.47(s,18h);如圖7所示。
在結構確認后,進行了熱化學性質測試,測試結果如下:初始揮發溫度為182℃,t50(失重50%時的溫度)為207℃,終止揮發溫度為225℃,揮發過程為單階揮發曲線、無多余拐點,且最終殘余質量僅為2%,說明(n,n’-二仲丁基-甲基脒基)(六甲基二硅氮基)ge(ⅱ)具有良好的揮發性及熱穩定性,適用于ald前驅體。
實施例6:式v化合物的制備及結構確認
100mlschlenk瓶抽真空充氬氣置換三次,氬氣保護下加入n-仲丁基-n’-異丙基-甲基脒基配體(0.468g,3mmol)、重蒸除水的乙醚(15ml),體系為無色溶液。在-78℃下,逐滴滴加正丁基鋰(1.2ml,3mmol)。緩慢恢復室溫,并繼續攪拌3h,體系為淡黃色溶液。
另取100mlschlenk瓶抽真空充氬氣置換三次,氬氣保護下加入二氯化鍺二氧六環(0.693g,3mmol)、重蒸除水的乙醚(10ml)。在氬氣氣體保護、-78℃下,將鋰鹽的乙醚溶液逐滴滴加至二氯化鍺二氧六環的乙醚液中。緩慢恢復室溫后并繼續攪拌12h。
另取100mlschlenk瓶抽真空充氬氣置換三次,在氬氣氣體保護下,加入六甲基二硅氮烷(0.484g,3mmol)、重蒸除水的乙醚(15ml),體系為無色溶液。在-78℃下,逐滴滴加正丁基鋰(1.2ml,3mmol)。緩慢恢復室溫后繼續攪拌3h,體系為無色溶液。
在氬氣氣體保護、-78℃下,將上述鋰鹽的乙醚溶液逐滴滴加至上述單取代的鍺配合物的乙醚液中。緩慢恢復室溫后并繼續攪拌12h。真空條件除去體系中易揮發組份,并用15ml*2重蒸除水的正己烷提取產物。真空條件除去提取液中易揮發組份,得到淡黃色油狀粗產物。提純:真空條件、油浴溫度110℃下,用微量減壓蒸餾裝置提純得到無色油狀液態產物0.689g(產率約57%)。
其中,1hnmr(400mhz,c6d6)δ3.33(dt,j=12.7,6.4hz,1h),3.16–3.03(m,1h),1.60–1.42(m,2h),1.27(s,3h),1.21–1.04(m,9h),0.85(dt,j=44.3,7.4hz,3h),0.44(d,j=1.3hz,18h);如圖8所示。
在結構確認后,進行了熱化學性質測試,測試結果如下:初始揮發溫度為156℃,t50(失重50%時的溫度)為180℃,終止揮發溫度為199℃,揮發過程為單階揮發曲線、無多余拐點,且最終殘余質量僅為0.2%,說明(n,n’-二仲丁基-甲基脒基)(六甲基二硅氮基)ge(ⅱ)具有良好的揮發性及熱穩定性,適用于ald前驅體。
以上所述僅是本發明的優選實施方式,應當指出,對于本技術領域的普通技術人員來說,在不脫離本發明原理的前提下,還可以做出若干改進和潤飾,這些改進和潤飾也應視為本發明的保護范圍。
- 還沒有人留言評論。精彩留言會獲得點贊!