一種石墨相氮化碳量子點的制備方法與流程
本發明涉及光催化材料領域,尤其涉及一種石墨相的氮化碳量子點的制備方法。
背景技術:
石墨相氮化碳(g-C3N4) 作為一種新興的非金屬可見光催化劑得到了國內外廣泛的關注和研究。它是一種直接帶隙半導體,理想條件下室溫禁帶寬度為 2.7 eV 左右,因為具有與碳材料相似的層狀堆積結構和 sp2雜化的π共軛電子能帶結構,因此 g-C3N4被認為是最有可能代替碳材料的新型應用功能材料。
量子點的特殊結構導致了它具有表面效應 、量子尺寸效應 、介電限域效應和宏觀量子隧道效應 ,從而派生出與宏觀體系和微觀體系不同的低維物性 ,展現出許多不同于宏觀塊體材料的物理化學性質和獨特的發光特性。
技術實現要素:
本發明的目的在于提供一種石墨相氮化碳量子點的制備方法,本發明將g-C3N4和量子點的優點結合起來,制備出石墨相氮化碳量子點(CNQDs)。溶液中具有熒光性質的石墨相氮化碳量子點在給電子體或者供電子體的作用下會發生熒光猝滅,量子點的這種受到光而發生電子轉移的現象,使得量子點能夠在光轉換和相關應用中發揮重要作用,可以與多種材料復合形成異質結催化材料,具有十分可觀的應用價值。
本發明是通過以下步驟來實現的:
1、稱取40g硫脲均勻平鋪于坩堝底部,將坩堝置于馬弗爐中高溫煅燒,煅燒溫度為500℃,升溫速率3℃/min,煅燒時間為2h,得到黃色固體(石墨相氮化碳體);
2、黃色固體冷卻至室溫,將其研磨均勻,再次置于馬弗爐中高溫煅燒,煅燒溫度為520℃,升溫速率3℃/min,煅燒時間為2h,得到淡黃色片狀固體(石墨相氮化碳片狀);
3、取20—100 mg上述淡黃色片狀固體加入30—50ml水或30ml濃氨水,超聲波分散5—9 h,之后取上層較均勻液體轉移到50ml聚四氟乙烯反應釜中,在水熱溫度180—200度、水熱時間10-12 h,冷卻至室溫即可得到產品。
上述制備方法的石墨相氮化碳片取用量為25-35mg、水熱溫度為180度或200度、超聲波分散時間為5 h。
本發明通過控制水熱反應的溫度和時間,將石墨相氮化碳片 “切”成具有熒光效應的量子點。G-C3N4具有類似石墨的片層結構,片層以 3-三嗪環為基本結構單元。3-三嗪環中,C、N 原子均發生 sp2 軌道雜化,與石墨結構中的 C-C 不同,所有原子的 p 軌道相互重疊而形成離域π鍵,結構中每個碳氮鍵的鍵長和鍵能都相等,這是一種與苯環結構類似的有機環狀結構。根據Markus Antonietti 教授對 g-C3N4的結構的深入研究,七嗪環構成的g-C3N4結構中,環內的 C-N 鍵長 0.1316 nm,C-N-C 鍵角為 116.6°;環外的 C-N 鍵長0.1442nm,C-N-C-鍵角為 120.0°。七嗪環構成的 g-C3N4的聚合度更高,通過密度泛函理論(DFT)理論計算,縮聚反應的過程中,七嗪環構成的 g-C3N4的結構更穩定。
本發明的技術效果是:本發明將g-C3N4 sheet七嗪環結構破壞,得到納米級尺寸的量子點,該方法簡便易行,操作簡單,填補了量子點制備領域的空白,而且,相對于傳統的半導體量子點和有機染料,此方法制備的量子點水溶性好,化學惰性高,易于功能化,耐光漂白,低毒性,因此,該量子點在光解有機物、殺菌和生物標記等方面,特別是在環境保護方面,有著廣闊的應用前景。
附圖說明
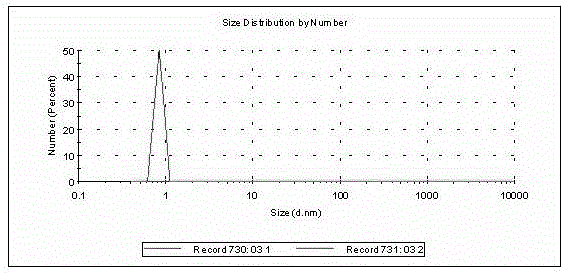
圖1為實施例1制得的產品粒徑測試結果圖。
圖2為實施例2制得的產品粒徑測試結果圖。
圖3為實施例3制得的產品粒徑測試結果圖。
圖4為實施例4制得的產品粒徑測試結果圖。
圖5為實施例5制得的產品粒徑測試結果圖。
圖6為實施例6制得的產品粒徑測試結果圖。
圖7為實施例7制得的產品粒徑測試結果圖。
圖8為實施例8制得的產品粒徑測試結果圖。
具體實施方式
下面將結合附圖1和實施例1-8來詳細說明本發明所具有的有益效果,旨在幫助閱讀者更好地理解本發明的實質,但不能對本發明的實施和保護范圍構成任何限定。
實施例1
取20mg 石墨相氮化碳片加入50ml水中,超聲波分散24h,取上層較均勻液體轉移到50ml聚四氟乙烯反應釜中,在水熱溫度200度、水熱時間10h進行反應,待冷卻至室溫即可得到產品。
實施例2
25mg 石墨相氮化碳片狀加入到30ml水,超聲波分散5h,用NaOH將溶液調節pH至12,之后取上層較均勻液體轉移到50ml聚四氟乙烯反應釜中,在水熱溫度200度、水熱時間10 h進行反應,待冷卻至室溫即可得到產品。
實施例3
50mg石墨相氮化碳片加入到30ml水,超聲波分散5h,用NaOH將溶液調節pH至12,之后取上層較均勻液體轉移到50ml聚四氟乙烯反應釜中,在水熱溫度200度、水熱時間10h 進行反應,待冷卻至室溫即可得到產品。
實施例4
100mg 石墨相氮化碳片加入到30ml水,超聲波分散5h,用NaOH將溶液調節pH至12,之后取上層較均勻液體轉移到50ml聚四氟乙烯反應釜中,在水熱溫度200度、水熱時間10h進行反應 ,待冷卻至室溫即可得到產品。
實施例5
25mg 石墨相氮化碳片加入到30ml濃氨水中,超聲波分散5h,之后取上層較均勻液體轉移到50ml聚四氟乙烯反應釜中,在水熱溫度180度、水熱時間12h進行反應,冷卻至室溫即可得到產品。
實施例6
35mg 石墨相氮化碳片加入到30ml濃氨水中,超聲波分散5h,之后取上層較均勻液體轉移到50ml聚四氟乙烯反應釜中,在水熱溫度180度、水熱時間12h進行反應,冷卻至室溫即可得到產品。
實施例7
25mg 石墨相氮化碳片加入到30ml濃氨水中,超聲波分散9h,之后取上層較均勻液體轉移到50ml聚四氟乙烯反應釜中,在水熱溫度180度、水熱時間12h進行反應,冷卻至室溫即可得到產品。
實施例8
35mg 石墨相氮化碳片加入到30ml濃氨水中,超聲波分散9h,之后取上層較均勻液體轉移到50ml聚四氟乙烯反應釜中,在水熱溫度180度、水熱時間12h進行反應,冷卻至室溫即可得到產品。
從上述實施例可知,不同的g-C3N4 sheet量對產品影響不大,不同超聲波分散時間對產品影響較大,超聲波分散時間過長反而效果不佳,水熱反應的溫度在180—200度效果最好。
綜上所述,本發明水熱法制備CNQDs時,石墨相氮化碳片量在25-35mg、水熱反應溫度200-180度、超聲波分散時間為5h,其技術效果最好。
以上所述的實施例僅僅是對本發明的優選實施方式進行描述,并非對本發明的范圍進行限定,在不脫離本發明設計精神的前提下,本領域普通技術人員對本發明的技術方案作出的各種變形和改進,均應落入本發明權利要求書確定的保護范圍內。
- 還沒有人留言評論。精彩留言會獲得點贊!