一種化合物在制備抑制激肽釋放酶KLK7的藥物上的應(yīng)用及其合成方法與流程
本發(fā)明屬于生物醫(yī)學(xué)領(lǐng)域,尤其是涉及一種化合物在制備抑制激肽釋放酶KLK7的藥物上的應(yīng)用及其合成方法。
背景技術(shù):
特應(yīng)性皮炎(Atopic Dermatitis,AD)是一種廣泛傳播的慢性炎癥性皮膚病,特點(diǎn)是干燥、鱗狀皮膚、炎癥、皮膚滲透性增加,AD病人的皮膚對(duì)環(huán)境中常見(jiàn)的而對(duì)正常人無(wú)害的因素也會(huì)很敏感從而易于受到表面感染。在過(guò)去的30年間工業(yè)化國(guó)家的特應(yīng)性皮炎的發(fā)病率增長(zhǎng)了2到3倍,據(jù)估計(jì)兒童的特應(yīng)性皮炎患病率達(dá)到了15-30%,成人的特應(yīng)性皮炎患病率達(dá)到了2-10%。由于發(fā)病機(jī)制的復(fù)雜性,對(duì)AD的發(fā)病機(jī)制了解得不多,但皮膚屏障功能紊亂已經(jīng)被證實(shí)是AD發(fā)展過(guò)程中的關(guān)鍵因素之一。皮膚屏障的穩(wěn)定依賴(lài)于顆粒層的角質(zhì)形成細(xì)胞分化為角質(zhì)細(xì)胞和皮膚表層角質(zhì)細(xì)胞脫落(即脫屑)之間的平衡,皮膚的脫屑過(guò)程包括由KLKs參與的橋粒的降解,由于橋粒是角質(zhì)細(xì)胞的粘連蛋白,橋粒的降解則會(huì)導(dǎo)致角質(zhì)細(xì)胞脫落而致皮膚脫屑。證據(jù)顯示,局部或暫時(shí)性的KLKs活性控制的失調(diào),是AD病人皮膚屏障穩(wěn)定缺失的主要原因之一。
KLKs(Kallikreins,激肽釋放酶)是具有胰蛋白酶或胰凝乳蛋白酶特異性的絲氨酸蛋白酶,其編碼基因定位于19q13.4,編碼15個(gè)分泌型絲氨酸蛋白酶KLK 1-KLK15。已經(jīng)證明,KLKs存在于正常人的角質(zhì)層細(xì)胞中,目前發(fā)現(xiàn),至少有8種KLK(KLK5,KLK 6,KLK 7,KLK 8,KLK10,KLK 11,KLK13和KLK14)存在于人的角質(zhì)層和汗液中,其中KLK7占總量的40%。KLK7可能是皮膚損傷的一個(gè)關(guān)鍵酶,而且在AD病人中它可能參與一個(gè)系統(tǒng)性炎癥反應(yīng),在AD的角質(zhì)層KLK7升高最為突出,這提示KLK7在AD的發(fā)病機(jī)制上可能是至關(guān)重要的,因此,抑制KLK7的活性對(duì)于AD的治療顯得最為重要。
LEKTI(Lympho-epithelial Kazal-type-related inhibitor,LEKTI)是由SPINK5基因編碼的絲氨酸蛋白酶抑制劑家族的成員,LEKTI前體包含一個(gè)信號(hào)肽和彼此隔開(kāi)的15個(gè)絲氨酸蛋白酶抑制域(D1-D15),表達(dá)后迅速被弗林蛋白酶水解為幾個(gè)蛋白片段,其中包含D6-D9抑制域的LEKTI能抑制胰蛋白酶、胰凝乳蛋白酶、KLK5和KLK7等酶的活性。LEKTI的內(nèi)源靶至少包括KLK5、KLK7和KLK14,它們共同參與皮膚的脫屑過(guò)程。在AD病人,LEKTI表達(dá)水平嚴(yán)重降低,導(dǎo)致了KLK7活性的異常升高。盡管LEKTI是表皮內(nèi)KLK7的天然抑制劑,但作為治療AD的外用藥,其不能透過(guò)皮膚角質(zhì)層,而且其大分子蛋白質(zhì)的特性也會(huì)導(dǎo)致吸收擴(kuò)散速度過(guò)慢、降解速度過(guò)快的問(wèn)題,因而作用非常有限。到目前為止,還沒(méi)有一種有效的KLK7抑制劑應(yīng)用于臨床。
技術(shù)實(shí)現(xiàn)要素:
基于此,本發(fā)明提供化合物在制備抑制激肽釋放酶KLK7的藥物上的應(yīng)用及其合成方法。
本發(fā)明技術(shù)方案為:
一種化合物在制備抑制激肽釋放酶KLK7的藥物上的應(yīng)用,該化合物的結(jié)構(gòu)式為:
合成所述的化合物的方法,所述合成路線(xiàn)如下:
其中:
物質(zhì)十二的合成:將物質(zhì)十、物質(zhì)十一、POCl3加入裝有回流冷凝管的圓底燒瓶中,將圓底燒瓶放置于超聲清洗儀中反應(yīng),減壓蒸出剩余的POCl3,殘余物傾倒于碎冰中,碎冰質(zhì)量為殘余物的20~50倍,析出沉淀,抽濾,固體用1%~5%的NaOH溶液洗滌,水洗至中性,得物質(zhì)十二;
物質(zhì)十三的合成:反應(yīng)瓶中加入二氯甲烷和物質(zhì)十二,鹽冰浴冷卻至-15℃,避光,氮?dú)獗Wo(hù)下,逐滴加入20wt%三溴化硼的無(wú)水二氯甲烷溶液,滴加完后,升溫反應(yīng),反應(yīng)結(jié)束后將反應(yīng)液倒入冰水中,冰水的質(zhì)量為反應(yīng)液的5-15倍,分出有機(jī)相,水相用乙酸乙酯萃取三次,每次乙酸乙酯的體積為水相的0.5~5倍,合并有機(jī)相,有機(jī)相用硫酸鈉干燥,蒸出溶劑,得物質(zhì)十三;
物質(zhì)十四的合成:將氯乙酸與5~20wt%氫氧化鈉水溶液混合得溶液A,將物質(zhì)十三與5~20wt%氫氧化鈉水溶液混合得溶液B,將溶液A加入到反應(yīng)瓶中,再滴加溶液B,滴加完畢,升溫反應(yīng),冷卻,加5~20wt%鹽酸酸化至pH=1~2,過(guò)濾、洗滌、干燥得物質(zhì)十四;
物質(zhì)十五的合成:在反應(yīng)瓶中裝有攪拌棒、溫度計(jì),回流冷凝管,其回流冷凝管連接氯化氫吸收裝置,依次加入氯化亞砜和物質(zhì)十四,攪拌升溫反應(yīng),減壓蒸出低沸物,瓶?jī)?nèi)殘余物即為物質(zhì)十五,不經(jīng)分離直接用于下步反應(yīng);
化合物II的合成:在反應(yīng)瓶中加入上步制得的物質(zhì)十五,再加入三乙胺、二氯甲烷和物質(zhì)十六,攪拌反應(yīng),減壓蒸出溶劑,冷卻,過(guò)濾,水洗,丙酮洗得化合物II。
優(yōu)選地,所述物質(zhì)十、物質(zhì)十一、POCl3的質(zhì)量比為1.0:1.0~1.5:30~50,超聲清洗儀中反應(yīng)溫度為50~70℃,功率為350W,超聲作用60~120分鐘。
優(yōu)選地,所述二氯甲烷、物質(zhì)十二、20wt%三溴化硼的二氯甲烷溶液的質(zhì)量比為20~40:1:6~15,升溫至25℃,25℃反應(yīng)2~4小時(shí)。
優(yōu)選地,所述溶液A中氯乙酸與氫氧化鈉的摩爾比為1:1.0~2.0,所述溶液B中物質(zhì)十三與氫氧化鈉的摩爾比為1:1.0~2.0,將溶液A加入到反應(yīng)瓶中,再滴加溶液B,加入到反應(yīng)瓶中的物質(zhì)十三與氯乙酸的摩爾比為1:1.0~2.0,升溫反應(yīng),冷卻。
優(yōu)選地,升溫至60~90℃反應(yīng)1~6小時(shí),冷卻至25℃。
優(yōu)選地,所述物質(zhì)十四與氯化亞砜的摩爾比為1:1.0~4.0,攪拌升溫至60~78℃,在60~78℃下反應(yīng)5~10小時(shí)。
優(yōu)選地,所述物質(zhì)十五、物質(zhì)十六、三乙胺和二氯甲烷的摩爾比1.0:1:0~2.0:1.0~3.0:5.0~30,在0~25℃攪拌2小時(shí)。
所述化合物或所述方法在制備抑制激肽釋放酶KLK7的藥物上的應(yīng)用,其特征在于,所述制備抑制激肽釋放酶KLK7的藥物上的應(yīng)用的藥物包括:
中的一種或幾種的混合物。
優(yōu)選地,所述制備抑制激肽釋放酶KLK7的藥物上的應(yīng)用的藥物包括:化合物I、化合物II、化合物III、化合物IV和化合物V的摩爾比為1:1~4:1~5:1~3:1~2。
本發(fā)明優(yōu)點(diǎn):
1、明提供的抑制激肽釋放酶KLK7的化合物能夠有效的抑制KLK7的活性,其合成方法簡(jiǎn)單易行,加入適當(dāng)?shù)妮o料和助劑可制備成抑制KLK7活性的藥物制劑。
2、采用本發(fā)明合成方法副產(chǎn)物產(chǎn)生的少,雜質(zhì)含量低至0.05%~0.08%。
3、本發(fā)明化合物和其他化合物聯(lián)用,對(duì)于激肽釋放酶KLK7的活性抑制效率更高。
4、在皮膚中主要表達(dá)的絲氨酸蛋白酶還包括KLK5,KLK14,Matriptase與Elastase2。本發(fā)明化合物合物對(duì)上述絲氨酸蛋白酶在100μM的濃度下均不變現(xiàn)出抑制活性,表明式其KLK7具有良好特異性。
5、本發(fā)明化合物在pH8,150mM NaCl,37℃的緩沖液中孵育72h,其對(duì)KLK7的抑制活性維持不變,表明本發(fā)明化合物的抑制效率高,活性強(qiáng),并且其抑制效果穩(wěn)定。
附圖說(shuō)明
圖1:本發(fā)明實(shí)施例式化合物II所示的濃度與抑制率的關(guān)系曲線(xiàn);
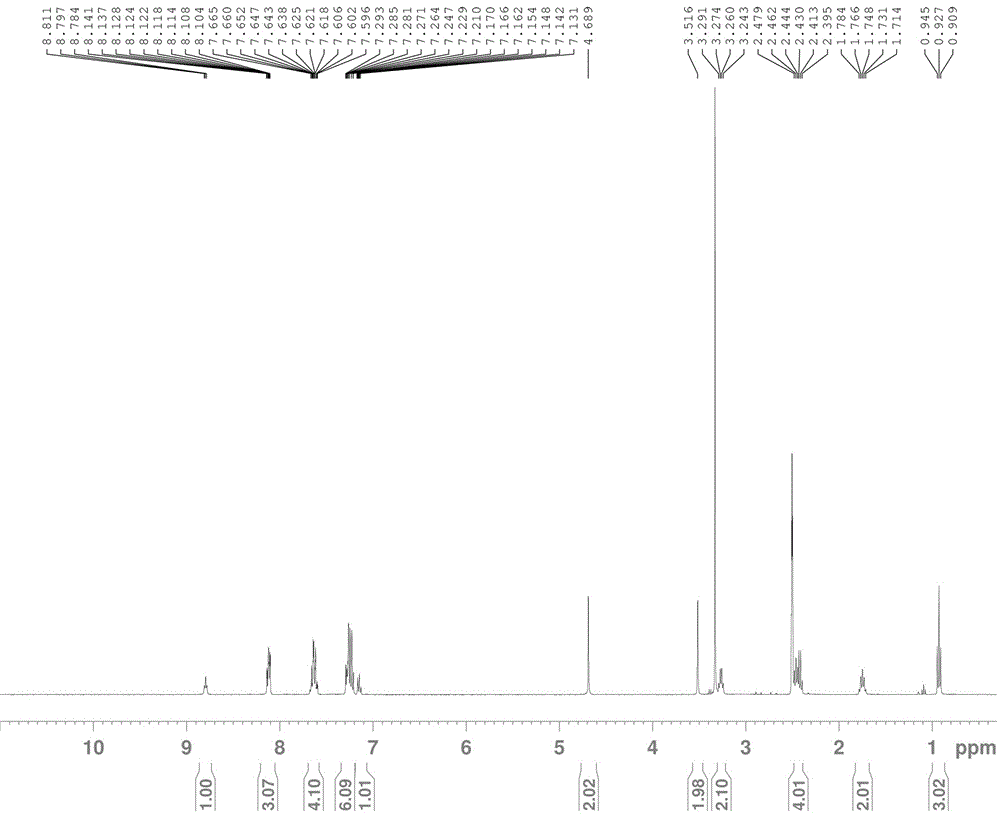
圖2:本發(fā)明化合物II的H1核磁圖;
圖3:本發(fā)明化合物II的C13核磁圖;
圖4:實(shí)施例7合化合物濃度與抑制率的關(guān)系曲線(xiàn)。
具體實(shí)施方式
下面結(jié)合實(shí)施例來(lái)進(jìn)一步說(shuō)明本發(fā)明,但本發(fā)明要求保護(hù)的范圍并不局限于實(shí)施例表述的范圍。
實(shí)施例1
化合物在制備抑制激肽釋放酶KLK7的藥物上的應(yīng)用,該化合物的結(jié)構(gòu)式為:
實(shí)施例2
合成實(shí)施例1所述的化合物的方法,所述合成路線(xiàn)如下:
其中:
物質(zhì)十二的合成:將物質(zhì)十、物質(zhì)十一、POCl3加入裝有回流冷凝管的圓底燒瓶中,將圓底燒瓶放置于超聲清洗儀中反應(yīng),減壓蒸出剩余的POCl3,殘余物傾倒于碎冰中,碎冰質(zhì)量為殘余物的20倍,析出沉淀,抽濾,固體用1%~5%的NaOH溶液洗滌,水洗至中性,得物質(zhì)十二;
物質(zhì)十三的合成:反應(yīng)瓶中加入二氯甲烷和物質(zhì)十二,鹽冰浴冷卻至-15℃,避光,氮?dú)獗Wo(hù)下,逐滴加入20wt%三溴化硼的無(wú)水二氯甲烷溶液,滴加完后,升溫反應(yīng),反應(yīng)結(jié)束后將反應(yīng)液倒入冰水中,冰水的質(zhì)量為反應(yīng)液的5倍,分出有機(jī)相,水相用乙酸乙酯萃取三次,每次乙酸乙酯的體積為水相的0.5倍,合并有機(jī)相,有機(jī)相用硫酸鈉干燥,蒸出溶劑,得物質(zhì)十三;
物質(zhì)十四的合成:將氯乙酸與5wt%氫氧化鈉水溶液混合得溶液A,將物質(zhì)十三與5wt%氫氧化鈉水溶液混合得溶液B,將溶液A加入到反應(yīng)瓶中,再滴加溶液B,滴加完畢,升溫反應(yīng),冷卻,加5wt%鹽酸酸化至pH=1,過(guò)濾、洗滌、干燥得物質(zhì)十四;
物質(zhì)十五的合成:在反應(yīng)瓶中裝有攪拌棒、溫度計(jì),回流冷凝管,其回流冷凝管連接氯化氫吸收裝置,依次加入氯化亞砜和物質(zhì)十四,攪拌升溫反應(yīng),減壓蒸出低沸物,瓶?jī)?nèi)殘余物即為物質(zhì)十五,不經(jīng)分離直接用于下步反應(yīng);
化合物II的合成:在反應(yīng)瓶中加入上步制得的物質(zhì)十五,再加入三乙胺、二氯甲烷和物質(zhì)十六,攪拌反應(yīng),減壓蒸出溶劑,冷卻,過(guò)濾,水洗,丙酮洗得化合物II。
所述物質(zhì)十、物質(zhì)十一、POCl3的質(zhì)量比為1:1:3,超聲清洗儀中反應(yīng)溫度為50℃,功率為350W,超聲作用60分鐘。
所述二氯甲烷、物質(zhì)十二、20wt%三溴化硼的二氯甲烷溶液的質(zhì)量比為20:1:6,升溫至25℃,25℃下反應(yīng)2小時(shí)。
所述溶液A中氯乙酸與氫氧化鈉的摩爾比為1:1,所述溶液B中物質(zhì)十三與氫氧化鈉的摩爾比為1:1,將溶液A加入到反應(yīng)瓶中,再滴加溶液B,加入到反應(yīng)瓶中的物質(zhì)十三與氯乙酸的摩爾比為1:1,升溫至60℃反應(yīng)1小時(shí),冷卻至25℃。
所述物質(zhì)十四與氯化亞砜的摩爾比為1:1,攪拌升溫至60℃,在60℃下反應(yīng)5小時(shí)。
所述物質(zhì)十五、物質(zhì)十六、三乙胺和二氯甲烷的摩爾比1:1:1:5,在0~25℃攪拌2小時(shí)。
實(shí)施例3
合成實(shí)施例1所述的化合物的方法,所述合成路線(xiàn)為實(shí)施例2所述:
物質(zhì)十二的合成:將物質(zhì)十、物質(zhì)十一、POCl3加入裝有回流冷凝管的圓底燒瓶中,將圓底燒瓶放置于超聲清洗儀中反應(yīng),減壓蒸出剩余的POCl3,殘余物傾倒于碎冰中,碎冰質(zhì)量為殘余物的50倍,析出沉淀,抽濾,固體用5%的NaOH溶液洗滌,水洗至中性,得物質(zhì)十二;
物質(zhì)十三的合成:反應(yīng)瓶中加入二氯甲烷和物質(zhì)十二,鹽冰浴冷卻至-15℃,避光,氮?dú)獗Wo(hù)下,逐滴加入20wt%三溴化硼的無(wú)水二氯甲烷溶液,滴加完后,升溫反應(yīng),反應(yīng)結(jié)束后將反應(yīng)液倒入冰水中,冰水的質(zhì)量為反應(yīng)液的15倍,分出有機(jī)相,水相用乙酸乙酯萃取三次,每次乙酸乙酯的體積為水相的0.5~5倍,合并有機(jī)相,有機(jī)相用硫酸鈉干燥,蒸出溶劑,得物質(zhì)十三;
物質(zhì)十四的合成:將氯乙酸與20wt%氫氧化鈉水溶液混合得溶液A,將物質(zhì)十三與20wt%氫氧化鈉水溶液混合得溶液B,將溶液A加入到反應(yīng)瓶中,再滴加溶液B,滴加完畢,升溫反應(yīng),冷卻,加20wt%鹽酸酸化至pH=2,過(guò)濾、洗滌、干燥得物質(zhì)十四;
物質(zhì)十五的合成:在反應(yīng)瓶中裝有攪拌棒、溫度計(jì),回流冷凝管,其回流冷凝管連接氯化氫吸收裝置,依次加入氯化亞砜和物質(zhì)十四,攪拌升溫反應(yīng),減壓蒸出低沸物,瓶?jī)?nèi)殘余物即為物質(zhì)十五,不經(jīng)分離直接用于下步反應(yīng);
化合物II的合成:在反應(yīng)瓶中加入上步制得的物質(zhì)十五,再加入三乙胺、二氯甲烷和物質(zhì)十六,攪拌反應(yīng),減壓蒸出溶劑,冷卻,過(guò)濾,水洗,丙酮洗得化合物II。
所述物質(zhì)十、物質(zhì)十一、POCl3的質(zhì)量比為1:1.5:50,超聲清洗儀中反應(yīng)溫度為70℃,功率為350W,超聲作用120分鐘。
所述二氯甲烷、物質(zhì)十二、20wt%三溴化硼的二氯甲烷溶液的質(zhì)量比為40:1:15,升溫至25℃,25℃下反應(yīng)4小時(shí)。
所述溶液A中氯乙酸與氫氧化鈉的摩爾比為1:2,所述溶液B中物質(zhì)十三與氫氧化鈉的摩爾比為1:2,將溶液A加入到反應(yīng)瓶中,再滴加溶液B,加入到反應(yīng)瓶中的物質(zhì)十三與氯乙酸的摩爾比為1:2,升溫至90℃反應(yīng)6小時(shí),冷卻至25℃。
所述物質(zhì)十四與氯化亞砜的摩爾比為1:4,攪拌升溫至78℃,在78℃下反應(yīng)10小時(shí)。
所述物質(zhì)十五、物質(zhì)十六、三乙胺和二氯甲烷的摩爾比1:2:3:30,在25℃攪拌2小時(shí)。
實(shí)施例4
合成實(shí)施例1所述的化合物的方法,所述合成路線(xiàn)為實(shí)施例2所述:
物質(zhì)十二的合成:將物質(zhì)十、物質(zhì)十一、POCl3加入裝有回流冷凝管的圓底燒瓶中,將圓底燒瓶放置于超聲清洗儀中反應(yīng),減壓蒸出剩余的POCl3,殘余物傾倒于碎冰中,碎冰質(zhì)量為殘余物的30倍,析出沉淀,抽濾,固體用3%的NaOH溶液洗滌,水洗至中性,得物質(zhì)十二;
物質(zhì)十三的合成:反應(yīng)瓶中加入二氯甲烷和物質(zhì)十二,鹽冰浴冷卻至-15℃,避光,氮?dú)獗Wo(hù)下,逐滴加入20wt%三溴化硼的無(wú)水二氯甲烷溶液,滴加完后,升溫反應(yīng),反應(yīng)結(jié)束后將反應(yīng)液倒入冰水中,冰水的質(zhì)量為反應(yīng)液的10倍,分出有機(jī)相,水相用乙酸乙酯萃取三次,每次乙酸乙酯的體積為水相的0.3倍,合并有機(jī)相,有機(jī)相用硫酸鈉干燥,蒸出溶劑,得物質(zhì)十三;
物質(zhì)十四的合成:將氯乙酸與15wt%氫氧化鈉水溶液混合得溶液A,將物質(zhì)十三與15wt%氫氧化鈉水溶液混合得溶液B,將溶液A加入到反應(yīng)瓶中,再滴加溶液B,滴加完畢,升溫反應(yīng),冷卻,加15wt%鹽酸酸化至pH=1,過(guò)濾、洗滌、干燥得物質(zhì)十四;
物質(zhì)十五的合成:在反應(yīng)瓶中裝有攪拌棒、溫度計(jì),回流冷凝管,其回流冷凝管連接氯化氫吸收裝置,依次加入氯化亞砜和物質(zhì)十四,攪拌升溫反應(yīng),減壓蒸出低沸物,瓶?jī)?nèi)殘余物即為物質(zhì)十五,不經(jīng)分離直接用于下步反應(yīng);
化合物II的合成:在反應(yīng)瓶中加入上步制得的物質(zhì)十五,再加入三乙胺、二氯甲烷和物質(zhì)十六,攪拌反應(yīng),減壓蒸出溶劑,冷卻,過(guò)濾,水洗,丙酮洗得化合物II。
所述物質(zhì)十、物質(zhì)十一、POCl3的質(zhì)量比為1:1.2:40,超聲清洗儀中反應(yīng)溫度為60℃,功率為350W,超聲作用100分鐘。
所述二氯甲烷、物質(zhì)十二、20wt%三溴化硼的二氯甲烷溶液的質(zhì)量比為30:1:10,升溫至25℃,25℃下反應(yīng)3小時(shí)。
所述溶液A中氯乙酸與氫氧化鈉的摩爾比為1:1.5,所述溶液B中物質(zhì)十三與氫氧化鈉的摩爾比為1:1.5,將溶液A加入到反應(yīng)瓶中,再滴加溶液B,加入到反應(yīng)瓶中的物質(zhì)十三與氯乙酸的摩爾比為1:1.5,升溫至80℃反應(yīng)4小時(shí),冷卻至25℃。
所述物質(zhì)十四與氯化亞砜的摩爾比為1:3,攪拌升溫至70℃,在70℃下反應(yīng)8小時(shí)。
所述物質(zhì)十五、物質(zhì)十六、三乙胺和二氯甲烷的摩爾比1:1:5:2:3,在20℃攪拌2小時(shí)。
實(shí)施例5
1)將化合物II溶解在DMSO中,配置成濃度10mM的母液;
2)在KLK7活性緩沖液中加入化合物II,配制成6.25μM,12.5μM,25μM,50μM,100μM,200μM的溶液,并在37℃作用15分鐘;
3)加入熒光底物MCa-R-P-K-P-V-E-Nval-W-R-K(Dnp)-NH2,在37℃下測(cè)KLK7的活性15分鐘,采用熒光酶標(biāo)儀檢測(cè)熒光值,根據(jù)熒光值的大小計(jì)算KLK7的活性。
陰性對(duì)照組:在KLK7的活性緩沖液中加入DMSO,其它操作條件均與實(shí)驗(yàn)組相同。
根據(jù)上述實(shí)驗(yàn)組和陰性對(duì)照組獲得的KLK7的活性計(jì)算化合物II對(duì)KLK7的抑制率,計(jì)算抑制率的公式為%inhibition=(1-a/b)×100%,計(jì)算結(jié)果如表1所示;其中,a為待測(cè)樣品的酶活,b為陰性對(duì)照的酶活。
表1為化合物II在不同濃度下對(duì)KLK7活性的抑制率,根據(jù)表1所示的結(jié)果繪制化合物II的濃度與抑制率的關(guān)系圖,采用KaleidaGraph軟件進(jìn)行曲線(xiàn)擬合得到化合物II的濃度與抑制率的關(guān)系曲線(xiàn),如圖1所示。
由表1和圖1的結(jié)果可以看出,隨著化合物II濃度的增加,對(duì)KLK7活性的抑制率逐漸增高,式化合物II的濃度為200μM時(shí),對(duì)KLK7活性的抑制率達(dá)到100%。
因此,式化合物II對(duì)KLK7具有優(yōu)異的抑制效果,該化合物作為活性組分加入適當(dāng)?shù)妮o料和助劑可制備成抑制KLK7活性的藥物制劑。
化合物II在不同濃度下對(duì)KLK7活性的抑制率
由上述描述可以看出,本發(fā)明提供的抑制激肽釋放酶KLK7的化合物的制備方法簡(jiǎn)單易行,能夠有效的抑制KLK7的活性,加入適當(dāng)?shù)妮o料和助劑可制備成抑制KLK7活性的藥物制劑。
實(shí)施例6
所述制備抑制激肽釋放酶KLK7的藥物上的應(yīng)用的藥物包括:
所述化合物I、化合物II、化合物III、化合物IV和化合物V的質(zhì)量比為1:1~2:1~2.5:2~3:1~2。
實(shí)施例7
實(shí)施例6組合物的中按摩爾比為1:1:1:1:1混合后,按照實(shí)施例5的方法測(cè)試不同濃度下對(duì)KLK7活性的抑制率,結(jié)果見(jiàn)表2和圖4
表2混合物在不同濃度下對(duì)KLK7活性的抑制率
由表2結(jié)果證明,式化合物I、化合物II、化合物III、化合物IV和化合物V聯(lián)用效果要比單獨(dú)使用效果更加顯著,同樣的添加量情況下,混合物的藥效要比單獨(dú)使用時(shí)對(duì)KLK7活性抑制率更高。
實(shí)施例8
合成化合物I的方法,所述合成路線(xiàn)如下:
其中:
物質(zhì)三的合成:向反應(yīng)瓶中加入乙醇、物質(zhì)一、質(zhì)量濃度37%的甲醛水溶液、氫氧化鈉以及物質(zhì)二,反應(yīng),反應(yīng)完畢,乙酸乙酯萃取三次,每次乙酸乙酯的用量為前述乙醇體積的1~5倍,合并有機(jī)相,蒸餾,冷卻,過(guò)濾,水洗,乙醇洗,干燥得物質(zhì)三;
物質(zhì)四的合成:向反應(yīng)瓶中加入物質(zhì)三、醋酐,控制溫度為-12℃,滴加質(zhì)量濃度為90%的發(fā)煙硝酸反應(yīng),反應(yīng)完畢,升至25℃,倒入冰水中,冰水的體積為反應(yīng)液5-10倍,過(guò)濾,先用1~5%碳酸氫鈉溶液洗,再用冰水洗,干燥得物質(zhì)四;
物質(zhì)五的合成:向反應(yīng)瓶中加入乙醇、水、醋酸、鐵粉、氯化銨和物質(zhì)四,回流反應(yīng),再加入乙醇,第二次加入乙醇的質(zhì)量為物質(zhì)四的5倍,趁熱過(guò)濾,濾液冷卻至25℃,倒入冰水中,冰水的體積為濾液的10倍,過(guò)濾,水洗,干燥得物質(zhì)五;
物質(zhì)七的合成:向反應(yīng)瓶中加入物質(zhì)五、物質(zhì)六、二甲基甲酰胺、碳酸鉀,反應(yīng),冷卻至25℃,將反應(yīng)液加入到冰水中,冰水的質(zhì)量為反應(yīng)液的10倍,過(guò)濾,水洗,干燥得物質(zhì)七。
物質(zhì)八的合成:向反應(yīng)瓶中加入物質(zhì)七和吡啶的混合物,再滴加三氯氧磷,升溫反應(yīng),將反應(yīng)液冷卻,加入氯仿,攪拌,冷卻,加入冷水,分出有機(jī)相,冷水洗氯仿層為中性,濃縮除去氯仿,干燥得物質(zhì)八。
化合物I的合成:將二甲基甲酰胺、物質(zhì)八、碳酸鉀和物質(zhì)九依次加到反應(yīng)瓶中,加熱反應(yīng),冷卻,將反應(yīng)液倒入水中,抽濾,濾餅用水淋洗,干燥得化合物I。
所述物質(zhì)二、質(zhì)量濃度37%的甲醛水溶液、物質(zhì)一、氫氧化鈉和乙醇的質(zhì)量比為1:1:1:1:5,25℃下反應(yīng)2時(shí)。
所述物質(zhì)三、醋酐、硝酸的質(zhì)量比為1:0.5:0.5,在-5℃下反應(yīng)4小時(shí)。
所述物質(zhì)四、乙醇、水、醋酸、鐵粉、氯化銨質(zhì)量比為1:2.5:2:0.05:1:0.02,回流反應(yīng)2小時(shí)。
所述物質(zhì)五、物質(zhì)六、碳酸鉀、二甲基甲酰胺的摩爾比為1:1:1:10,升溫至80℃反應(yīng)4小時(shí)。
所述物質(zhì)七、吡啶、三氯氧磷的摩爾比為1:1:1.5,加畢,升溫至100℃反應(yīng)3小時(shí),將反應(yīng)液冷卻至50℃,加入氯仿,氯仿的體積為吡啶的40倍,攪拌1小時(shí),冷卻至0℃,加入5℃的冷水,冷水的體積為氯仿的0.6倍,分出有機(jī)相,冷水洗氯仿層為中性,濃縮除去氯仿,干燥得物質(zhì)八。
所述物質(zhì)八、物質(zhì)九、碳酸鉀、二甲基甲酰胺的摩爾比為1:2:2:20,加熱至50℃,反應(yīng)1h,冷卻,將反應(yīng)液倒入水中,水的體積為反應(yīng)液的10倍,抽濾,濾餅用水淋洗,干燥得化合物I。
實(shí)施例9
合成化合物III的方法,所述合成路線(xiàn)如下:
其中:
物質(zhì)十八的合成:將物質(zhì)十七、KOH、水、二硫化碳加入到反應(yīng)瓶,,加熱至回流,反應(yīng)2小時(shí),冷至25℃,過(guò)濾,濾液用1wt%鹽酸調(diào)節(jié)pH值析出物質(zhì)十八,過(guò)濾、洗滌、干燥得物質(zhì)十八;
物質(zhì)二十的合成:向反應(yīng)瓶中加入物質(zhì)十八、物質(zhì)十九、甲苯和碳酸鉀,回流反應(yīng),冷卻過(guò)濾,用二氯甲烷洗固體,濾液減壓濃縮,殘留物用硅膠層析,己烷-乙酸乙酯洗脫,得物質(zhì)二十。
化合物III的合成:向反應(yīng)瓶中加入物質(zhì)二十、4-氟芐氯、NaOH、乙腈,回流反應(yīng),冷卻,減壓蒸出溶劑得殘留物,殘留物用乙酸乙酯萃取三次,每次乙酸乙酯的體積為殘留物的2倍,有機(jī)相水洗,再用無(wú)水硫酸鈉干燥,過(guò)濾,濾液減壓蒸出溶劑得粗品,粗品以乙腈重結(jié)晶得化合物III。
所述物質(zhì)十七、二硫化碳、KOH、水的摩爾比為1:1:1:5。
所述物質(zhì)十八、物質(zhì)十九、碳酸鉀、甲苯的摩爾比為1:1:1:5,回流反應(yīng),冷卻,所述己烷-乙酸乙酯體積比為2:1。
所述物質(zhì)二十合成中回流反應(yīng)時(shí)間為4小時(shí),冷卻至25℃。
所述物質(zhì)二十、4-氟芐氯、NaOH、乙腈的摩爾比為1:1:1:10,回流反應(yīng)4小時(shí)。
實(shí)施例10
合成化合物Ⅳ的方法,所述合成路線(xiàn)如下:
其中:
物質(zhì)二十三的合成:將物質(zhì)二十一、物質(zhì)二十二、冰醋酸加入到反應(yīng)瓶,加熱至回流反應(yīng)4-~8小時(shí),冷卻,過(guò)濾,水洗,二氯甲烷洗,得物質(zhì)二十三;
物質(zhì)二十四的合成:向反應(yīng)瓶中加入物質(zhì)二十三和吡啶的混合物,再滴加三氯氧磷,加畢,升溫反應(yīng),反應(yīng)結(jié)束后將反應(yīng)液冷卻至50℃,加入氯仿,攪拌反應(yīng),冷卻至5℃,加入冷水,分出有機(jī)相,冷水洗氯仿層為中性,濃縮除去氯仿,干燥得物質(zhì)二十四;
化合物Ⅳ的合成:向反應(yīng)瓶中加入物質(zhì)二十四、物質(zhì)二十五、甲苯、碳酸鉀,回流反應(yīng)4小時(shí),冷至25℃,過(guò)濾,用二氯甲烷洗固體,濾液減壓濃縮得粗品,粗品以氯仿重結(jié)晶得化合物Ⅳ。
所述物質(zhì)二十一、物質(zhì)二十二、冰醋酸的摩爾比為1:1:1.5,回流反應(yīng)4小時(shí),冷卻至25℃。
所述物質(zhì)二十三、吡啶、三氯氧磷的摩爾比為1:1:1.5,升溫至110℃反應(yīng)2小時(shí),將反應(yīng)液冷卻至50℃,加入氯仿,所述氯仿的體積為吡啶的30倍,攪拌反應(yīng)1小時(shí),冷卻至0℃,加入5℃的冷水,冷水的體積為氯仿的0.5倍。
實(shí)施例11
合成化合物V的工藝,所述合成路線(xiàn)如下:
其中:
物質(zhì)二十八的合成:將物質(zhì)二十六和物質(zhì)二十七加入干燥圓底燒瓶中,升溫進(jìn)行攪拌反應(yīng),薄層色譜跟蹤反應(yīng)至原料消失,將混合物冷卻至25℃,倒入冰水混合物中,過(guò)濾,水洗,氯仿洗,得物質(zhì)二十八;
物質(zhì)二十九的合成:反應(yīng)瓶中加入依次乙醚、氫化鋰鋁,攪拌溶解,冰水浴冷卻至0℃,再緩慢加入物質(zhì)二十八,加料完畢,冰水浴下反應(yīng),將反應(yīng)液倒入冰水中,靜置讓沉淀物停在底部,過(guò)濾,氯仿洗濾渣,濾液分出有機(jī)相,水相以氯仿萃取三次,有機(jī)相合并,減壓濃縮得物質(zhì)二十九。
化合物V的合成:在反應(yīng)中加入物質(zhì)二十九、三乙胺、二氯甲烷和物質(zhì)三十,攪拌反應(yīng),濃縮除去二氯甲烷,然后加水,加水質(zhì)量為濃縮物的10倍,用氯仿萃取三次,萃取劑氯仿每次用量為前述所加水的體積的1倍,有機(jī)相合并,減壓濃縮得粗品,粗品用氯仿重結(jié)晶得化合物V。
所述物質(zhì)二十六、物質(zhì)二十七的摩爾比為1:1.,升至80℃進(jìn)行攪拌反應(yīng),所述冰水混合物的質(zhì)量為物質(zhì)二十六的5倍。
所述物質(zhì)二十八、氫化鋰鋁、乙醚的質(zhì)量比為1:0.5:15,加料速度以微回流為宜,所述冰水浴下反應(yīng)1.5小時(shí),將反應(yīng)液倒入冰水中,冰水的體積為反應(yīng)液的1倍,萃取劑氯仿每次用量為水相體積的1倍。
所述物質(zhì)二十九、物質(zhì)三十、三乙胺和二氯甲烷的摩爾比為1:1:1:10,25℃攪拌1.5小時(shí)。
上述的實(shí)施例僅為本發(fā)明的優(yōu)選技術(shù)方案,而不應(yīng)視為對(duì)于本發(fā)明的限制,本申請(qǐng)中的實(shí)施例及實(shí)施例中的特征在不沖突的情況下,可以相互任意組合。本發(fā)明的保護(hù)范圍應(yīng)以權(quán)利要求記載的技術(shù)方案,包括權(quán)利要求記載的技術(shù)方案中技術(shù)特征的等同替換方案為保護(hù)范圍。即在此范圍內(nèi)的等同替換改進(jìn),也在本發(fā)明的保護(hù)范圍之內(nèi)。
- 還沒(méi)有人留言評(píng)論。精彩留言會(huì)獲得點(diǎn)贊!
- 化合物在制備抑制激肽釋放酶KLK7的藥物上的應(yīng)用及其合成工藝的制作方法與工藝
- 新的激肽釋放酶7 抑制劑的制作方法與工藝
- 用天冬酰胺內(nèi)肽酶(AEP)抑制劑以及相關(guān)組合物對(duì)神經(jīng)退行性疾病的治療的制作方法與工藝
- 用作乙酰輔酶 A 羧化酶抑制劑類(lèi)的吡唑并螺酮衍生物的制作方法與工藝
- BACE1的肽抑制劑的制造方法與工藝
- 一種羧肽酶A固定化酶的制備方法與流程
- 作為血漿激肽釋放酶抑制劑的N?((雜)芳基甲基)?雜芳基?甲酰胺化合物的制造方法與工藝
- 改良的鈉通道的肽抑制劑的制造方法與工藝
- 一種血管緊張素轉(zhuǎn)化酶抑制肽及其制備提取方法與流程
- 一種利用魚(yú)鱗制備ACE抑制肽的方法與制造工藝
- 新的激肽釋放酶7 抑制劑的制作方法與工藝
- 用天冬酰胺內(nèi)肽酶(AEP)抑制劑以及相關(guān)組合物對(duì)神經(jīng)退行性疾病的治療的制作方法與工藝
- 一種新生血管生成抑制肽及其透明質(zhì)酸修飾物的制備方法與應(yīng)用與流程
- BACE1的肽抑制劑的制造方法與工藝
- 作為血漿激肽釋放酶抑制劑的N?((雜)芳基甲基)?雜芳基?甲酰胺化合物的制造方法與工藝
- 改良的鈉通道的肽抑制劑的制造方法與工藝
- 一種血管緊張素轉(zhuǎn)化酶抑制肽及其制備提取方法與流程
- 一種利用魚(yú)鱗制備ACE抑制肽的方法與制造工藝
- 作為ace和nep抑制劑的吡喃衍生物的制作方法
- 由蛇毒腺分泌的肽的藥物組合物制劑的制作方法
- 鑒定改變iRhom多肽活性的化合物的方法及其用途與流程
- 鑒定改變iRhom多肽活性的化合物的方法及其用途與流程
- 一種高純度環(huán)己肽類(lèi)化合物的制備方法與流程
- 一種新型環(huán)肽化合物在制備抑制結(jié)核桿菌的藥物中的應(yīng)用的制作方法與工藝
- 化合物在制備抑制激肽釋放酶KLK7的藥物上的應(yīng)用及其合成工藝的制作方法與工藝
- 一種環(huán)肽類(lèi)抗真菌化合物及其制備方法與流程
- 一種環(huán)肽類(lèi)化合物及其制備方法和應(yīng)用與流程
- 環(huán)肽類(lèi)化合物及其制備和用途的制作方法與工藝
- 擬肽大環(huán)化合物及其用途的制造方法與工藝
- 肽化合物、包含它們的組合物和所述化合物,特別是化妝品的用途的制造方法與工藝
- 一種酶催化合成谷胱甘肽的方法
- Nannocystin A及其結(jié)構(gòu)類(lèi)似物的合成的制作方法
- 一種石墨烯信號(hào)放大的微囊藻毒素?lr電化學(xué)檢測(cè)方法
- 一種西蘭花多肽組分微波輔助提取及活性測(cè)定方法
- 一種鐮孢菌菌劑、含脂肽類(lèi)化合物的農(nóng)藥及用圖
- 二肽類(lèi)化合物的鹽酸鹽及其制備方法
- 含有脂質(zhì)肽型化合物的棒狀基材的制作方法
- 一種通過(guò)分步補(bǔ)料提高l-纈氨酸產(chǎn)酸的發(fā)酵工藝的制作方法
- 一種四氫吲哚-4酮三肽類(lèi)化合物,制備方法及其在抗腫瘤藥物中的用圖
- 一種通過(guò)分步補(bǔ)料提高l-纈氨酸產(chǎn)酸的發(fā)酵工藝的制作方法