石蒜科植物忽地笑細胞色素P450還原酶1及其編碼基因與應用的制作方法
本發明涉及生物技術和植物生物學領域;更具體地,本發明涉及一種來源于石蒜科植物的細胞色素P450還原酶及其編碼基因與應用。
背景技術:
植物細胞色素P450酶(Cytochrome P450enzymes,CYPs)參與大量植物天然產物的生物合成,是天然產物生物合成的關鍵酶之一,屬于亞鐵血紅素單加氧酶超大家族,催化多種類型的氧化還原反應,具有結構選擇性、區域選擇性和立體選擇性。CYPs的催化活性嚴格依賴于細胞色素P450還原酶。細胞色素P450還原酶(EC1.6.2.4Cytochrome P450Reductase,CPR)是CYPs催化體系的重要組成部分,屬于核黃素蛋白家族,具有黃素腺嘌呤二核苷酸(FAD)結合結構域和黃素單核苷酸(FMN)結合結構域。作為CYPs的氧化還原分子伴侶,細胞色素P450還原酶可以從電子供體(還原力)中獲得電子,并通過輔因子FAD和FMN,將獲得的電子傳遞給CYPs,協助CYPs所催化的一系列氧化還原反應。如果沒有CPR的存在,CYPs對其底物就沒有催化活性。因此,CPR是CYPs催化進行結構專一性、區域專一性和立體專一性生物反應所必需的:CPR催化電子從電子供體(還原力)通過FAD和FMN傳遞到CYPs的亞鐵血紅素輔基,協助CYPs催化底物的生物轉化。
植物細胞中CYPs的種類數量一般有200多種,而CPR蛋白卻只有2-5種。也就是說,植物CPR蛋白在協助植物CYPs進行底物的轉化(產物的合成)過程中具有較好的適用性。因此,在研究細胞色素P450酶的催化活性以及利用細胞色素P450酶進行生物轉化和生物合成時,如果沒有對應物種的CPR,可以使用來源于其他物種的CPR代替。也就是說,CPR在植物天然產物的體內合成、生物轉化以及發酵過程中具有重要的作用。
忽地笑(Lycoris aurea)是多年生草本球根藥用植物,屬于石蒜科石蒜屬,富含加蘭他敏、石蒜堿、文殊蘭堿等石蒜科植物所特有的生物堿(石蒜科生物堿)與其它生物堿、以及其它類型的植物天然產物。這些天然產物具有重要的生物活性和應用價值。例如,加蘭他敏作為一種特定的、競爭性的、可逆的乙酰膽堿酯酶抑制劑,在臨床上用于治療阿爾茨海默病(老年癡呆癥)。又例如,作為一種苯酚衍生物,對-香豆酸是植物苯丙素類天然產物,具有抗氧化、抗病毒、抗癌及抗炎等多種生物醫藥活性,用于臨床及醫藥、食品和化妝品等行業。作為一個多功能的藥效基團和平臺化合物,對-香豆酸還可以衍生出大量具有醫藥活性的化合物(包括上述在臨床治療輕、中度老年癡呆癥的藥物-加蘭他敏等),具有廣泛的應用和需求。在這些天然產物的生物合成過程中,細胞色素P450酶(CYPs)參與催化一步或多步反應,而其催化功能依賴于細胞色素P450還原酶(CPR)。但是,石蒜屬植物細胞色素P450還原酶及其編碼基因尚未被分離與克隆。因此,本領域有必要開發石蒜屬植物忽地笑的細胞色素P450還原酶。該蛋白及其編碼基因可通過植物轉基因和異源表達的方式協助細胞色素P450酶進行天然產物的生物轉化與生物合成。
技術實現要素:
本發明的目的在于提供一種石蒜科植物的細胞色素P450還原酶,所述的細胞色素P450還原酶選自:
(a)氨基酸序列如SEQ ID NO:1所示的蛋白質;或
(b)將SEQ ID NO:1氨基酸序列經過一個或多個(如1-90個)氨基酸殘基的取代、缺失或添加而形成的,且具有細胞色素P450還原酶活性的由(a)衍生的蛋白質;或
(c)與SEQ ID NO:1氨基酸序列有至少86%序列相同性,且具有細胞色素P450還原酶活性的由(a)衍生的蛋白質。
本發明SEQ ID NO:1所示的蛋白質是從忽地笑(Lycoris aurea)中分離得到的一種新的細胞色素P450還原酶。為便于表述,將SEQ ID NO:1所示的蛋白質命名為細胞色素P450還原酶LaCPR1。
在一個優選例中,所述的細胞色素P450還原酶活性是指利用還原型煙酰胺腺嘌呤二核苷酸磷酸(NADPH)為電子供體(還原力),傳遞電子給細胞色素P450酶的替代物(細胞色素c)。
在另一個優選例中,所述的細胞色素P450還原酶活性是指傳遞電子給細胞色素P450酶(Cytochrome P450enzyme,CYPs),并協助CYPs催化其底物合成產物;例如植物天然產物的生物轉化與生物合成。
在另一個優選例中,所述的序列(c)還包括:由(a)或(b)添加了標簽序列、信號序列或分泌信號序列后所形成的融合蛋白。
本發明的另一目的在于提供分離的多核苷酸,該多核苷酸是編碼所述細胞色素P450還原酶的多核苷酸。
在一個優選例中,該多核苷酸的核苷酸序列如SEQ ID NO:2所示。
應理解,考慮到密碼子的簡并性以及不同物種密碼子的偏好性,本領域技術人員可以根據需要使用合適特定物種表達的密碼子。因而,本發明細胞色素P450還原酶的多核苷酸還包括由SEQ ID NO:2所示核苷酸序列經取代、缺失和/或增加一個或幾個核苷酸,得到的編碼具有細胞色素P450還原酶活性的核苷酸序列。
本發明的又一目的是提供一種載體,它含有所述的多核苷酸。所述的載體是將本發明的編碼所述細胞色素P450還原酶的多核苷酸與表達載體可操作地連接,得到能夠表達本發明所述細胞色素P450還原酶的重組表達載體或抑制本發明所述細胞色素P450還原酶編碼多核苷酸表達的基因沉默載體。
在一個優選例中,該載體是含有編碼所述細胞色素P450還原酶的SEQ ID NO:2所示序列的重組表達載體pET28a-LaCPR1。
在另一個優選例中,該載體是含有編碼所述細胞色素P450還原酶的SEQ ID NO:2所示序列中第175-2088位多核苷酸的重組表達載體pET28a-LaCPR1(ΔN58)。
本發明的又一目的是提供一種宿主細胞,它含有所述的載體或基因組中整合有所述的多核苷酸。所述的宿主細胞是原核細胞或真核細胞。常用的原核宿主細胞包括大腸桿菌、枯草桿菌、運動假單胞菌和乳酸菌等;常用的真核宿主細胞包括真菌細胞、植物細胞、昆蟲細胞和哺乳動物細胞等。所述的真菌細胞包括酵母細胞。將所述的重組表達載體或者基因沉默載體導入所述的適當宿主細胞中,獲得表達本發明所述細胞色素P450還原酶的基因工程菌株、轉基因細胞系、轉基因愈傷、轉基因組織、轉基因植株或者基因工程植株。
本發明的又一目的是提供所述的細胞色素P450還原酶的用途,用于傳遞電子給細胞色素P450酶并協助細胞色素P450酶對其底物進行氧化修飾合成產物。
在一個優選例中,所述的細胞色素P450酶是CYP73A;或所述的底物是反式-肉桂酸。
本發明的又一目的是提供一種表達構建物。所述表達構建物包括以下酶的編碼基因和/或基因表達盒:
所述的細胞色素P450還原酶;和
細胞色素P450酶;和/或
細胞色素P450酶底物的生物合成酶。
所述基因表達盒是酶在宿主細胞中表達與調控所需要的生物學元件,包括啟動子、增強子、衰減子、核糖體結合位點、Kozak序列、內含子和/或轉錄終止子等;此外,還可包括標簽編碼序列和/或信號(肽)編碼序列等。
在一個優選例中,所述的表達構建物還包括:反式-肉桂酸-4-羥化酶(CYP73A)的編碼基因。
在另一個優選例中,所述的表達構建物還包括:反式-肉桂酸-4-羥化酶(CYP73A)的編碼基因和苯丙氨酸氨基裂解酶(PAL)編碼基因。
在另一個優選例中,當轉化大腸桿菌細胞時,所述的表達構建物中,還包含大腸桿菌啟動子、大腸桿菌核糖體結合位點和/或大腸桿菌轉錄終止子等基因表達盒。
本發明的又一目的是提供一種宿主細胞。所述的宿主細胞中包括所述的表達構建物。所述的宿主細胞是原核細胞或真核細胞。常用的原核宿主細胞包括大腸桿菌、枯草桿菌、運動假單胞菌和乳酸菌等;常用的真核宿主細胞包括真菌細胞、植物細胞、昆蟲細胞和哺乳動物細胞等。較佳地,所述的宿主細胞是內源存在細胞色素P450酶的底物或其底物前體的細胞。
本發明的又一目的是提供所述的表達構建物的用途,用于生產對-香豆酸。
本發明的又一目的是提供一種生產對-香豆酸的方法。所述方法包括:利用所述的細胞色素P450還原酶作為氧化還原分子伴侶,協助細胞色素P450酶CYP73A催化反式-肉桂酸合成對-香豆酸。
在一個優選例中,所述方法包括:以所述的表達構建物轉化宿主細胞,利用轉化的宿主細胞催化反式-肉桂酸合成對-香豆酸;所述的宿主細胞是原核細胞或真核細胞。常用的原核宿主細胞包括大腸桿菌、枯草桿菌、運動假單胞菌和乳酸菌等;常用的真核宿主細胞包括真菌細胞、植物細胞、昆蟲細胞和哺乳動物細胞等。所述的真菌細胞包括酵母細胞。
在另一個優選例中,所述方法包括:以所述的表達構建物轉化宿主細胞,培養轉化的大腸桿菌細胞,利用簡單的碳水化合物(葡萄糖)合成對-香豆酸。該宿主細胞是包含有細胞色素P450酶的底物的細胞;較佳地,該宿主細胞是內源存在反式-肉桂酸或其前體的細胞。
本發明首次揭示了石蒜科植物忽地笑來源的細胞色素P450還原酶LaCPR1,其具有良好的輔酶專一性。本發明還揭示了編碼所述細胞色素P450還原酶的多核苷酸、表達所述細胞色素P450還原酶的表達載體及宿主細胞。本發明應用石蒜科植物來源的細胞色素P450還原酶,可協助細胞色素P450酶(CYPs)發揮催化活性,進而實現天然產物(例如但不限于對-香豆酸)的生物轉化以及生物合成。
附圖說明
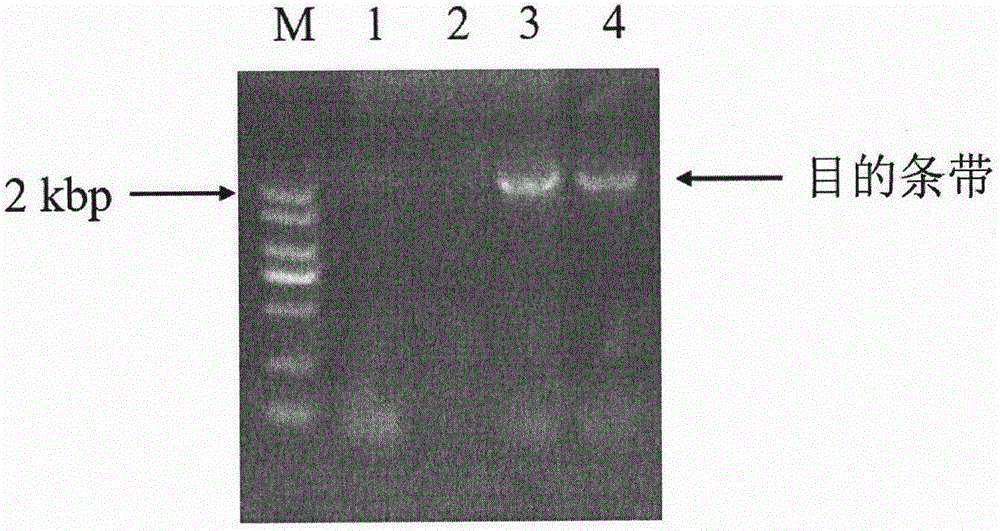
圖1是引物對SEQ ID NO:3和SEQ ID NO:4的聚合酶鏈式反應(PCR)擴增產物的瓊脂糖凝膠電泳檢測結果。
圖2是重組表達載體pET28a-LaCPR1的菌落PCR驗證電泳圖(引物為T7和T7ter)。
圖3是重組表達載體pET28a-LaCPR1(ΔN58)的菌落PCR驗證電泳圖(引物為T7和T7ter)。
圖4是LaCPR1在大腸桿菌中表達與分布檢測的SDS-PAGE電泳圖。
圖5是LaCPR1的輔因子專一性與酶活性圖。
圖6是LaCPR1(ΔN58)在大腸桿菌中表達與分布檢測的SDS-PAGE電泳圖。
圖7是LaCPR1(ΔN58)的輔因子專一性與酶活性圖。
圖8是重組表達載體pET28a-LaCPR1-CYP73A的菌落PCR驗證電泳圖(引物為SEQ ID NO:10和T7ter)。
圖9是重組表達載體pET28a-LaCPR1(ΔN58)-CYP73A的菌落PCR驗證電泳圖(引物為SEQ ID NO:10和T7ter)。
圖10是重組表達載體pET28a-CYP73A的菌落PCR驗證電泳圖(引物為T7和T7ter)。
圖11是重組菌株EcR-LaCPR1、EcR-CYP73A、EcR-LaCPR1-CYP73A和EcR-LaCPR1(ΔN58)-CYP73A生物轉化圖。
圖12是重組表達載體pET28a-LaCPR1(ΔN58)-CYP73A-rbsPAL的菌落PCR驗證電泳圖(引物為SEQ ID NO:13和T7ter)。
圖13是重組菌株Ec31884-LaCPR1(ΔN58)-CYP73A-PAL發酵生產對-香豆酸的產量圖。
具體實施方式
以下結合具體實施例并附圖,進一步闡述本發明。
以下實施例進一步說明本發明的內容,但不應理解為對本發明的限制。在不背離本發明精神和實質的情況下,對本發明方法、步驟或條件所作的修改或替換,均屬于本發明的范圍。
若未特別指明,實施例中所用的技術手段為本領域技術人員所熟知的常規手段。
實施例1、細胞色素P450還原酶LaCPR1編碼基因的克隆
合成兩條引物分別具有序列表中SEQ ID NO:3、SEQ ID NO:4的核苷酸序列。
以從忽地笑中提取的RNA反轉錄獲得的cDNA為模板,利用如上兩條引物SEQ ID NO:3和SEQ ID NO:4進行PCR。DNA聚合酶選用南京諾唯贊生物科技有限公司的Super-Fidelity DNA聚合酶。PCR擴增程序為:95℃5min;94℃45s,56℃45s,72℃2min,共30個循環;72℃10min,降至10℃。PCR產物經瓊脂糖凝膠電泳檢測,結果如圖1。
在紫外燈照射下,切下目標DNA條帶。然后采用多功能DNA純化試劑盒(離心柱型)(北京百泰克生物技術有限公司)從瓊脂糖凝膠中回收DNA即為擴增出的細胞色素P450還原酶編碼基因的DNA片段。利用寶生物工程(大連)有限公司(TaKaRa)的pMD19-T克隆試劑盒,將回收的PCR產物克隆到pMD19-T載體,所構建的載體命名為pMDT-LaCPR1。經測序獲得LaCPR1的基因序列。
LaCPR1基因具有序列表中SEQ ID NO:2的核苷酸序列。自SEQ ID NO:2的5’-端第1-2088位核苷酸為LaCPR1的開放閱讀框(Open Reading Frame,ORF),自SEQ ID NO:2的5’-端的第1-3位核苷酸為LaCPR1基因的起始密碼子ATG,自SEQ ID NO:2的5’-端的第2086-2088位核苷酸為LaCPR1基因的終止密碼子TGA。細胞色素P450還原酶編碼基因LaCPR1編碼一個含有695個氨基酸的蛋白質LaCPR1,具有SEQ ID NO:1的氨基酸序列,用軟件預測到該蛋白質的理論分子量大小為77.4KDa,等電點pI為5.27。
實施例2、LaCPR1基因的重組表達載體的構建
(1)合成分別具有序列表中SEQ ID NO:5和SEQ ID NO:7核苷酸序列的兩條引物。在合成的引物SEQ ID NO:5和SEQ ID NO:7的5’-端分別設置NdeI和XhoI兩個酶切位點及其保護堿基序列,以忽地笑的cDNA為模板進行PCR擴增。PCR擴增程序同實施例1。PCR擴增產物經瓊脂糖凝膠電泳檢測、分離、切膠回收后經NdeI和XhoI雙酶切,利用寶生物工程(大連)有限公司(TaKaRa)的T4DNA連接酶連接同樣經NdeI和XhoI雙酶切的pET28a載體(Novagen)中。連接產物轉化大腸桿菌(E.coli)DH5α(購自南京諾唯贊生物科技有限公司)感受態細胞,并涂布于添加25μg/mL卡那霉素的LB平板上。通過菌落PCR驗證獲得陽性轉化子,菌落PCR產物的瓊脂糖電泳結果如圖2。通過測序進一步驗證重組質粒pET28a-NdeI-LaCPR1-XhoI構建成功,并在NdeI和XhoI酶切位點之間含有SEQ ID NO:2的全長多核苷酸序列。所獲得的重組質粒命名為pET28a-LaCPR1。
(2)合成分別具有序列表中SEQ ID NO:6和SEQ ID NO:7核苷酸序列的兩條引物。在合成的引物SEQ ID NO:6和SEQ ID NO:7的5’-端分別設置NdeI和XhoI兩個酶切位點及其保護堿基序列,以忽地笑的cDNA為模板進行PCR擴增。PCR擴增程序同實施例1。PCR擴增產物經瓊脂糖凝膠電泳檢測、分離、切膠回收后經NdeI和XhoI雙酶切,利用T4DNA連接酶(購自寶生物工程(大連)有限公司(TaKaRa))連接同樣經NdeI和XhoI雙酶切的pET28a載體(Novagen)中。連接產物轉化大腸桿菌(E.coli)DH5α(購自南京諾唯贊生物科技有限公司)感受態細胞,并涂布于添加卡那霉素(終濃度為25μg/mL)的LB平板上。通過菌落PCR驗證陽性轉化子,菌落PCR產物的瓊脂糖電泳結果如圖3。測序進一步驗證重組質粒pET28a-NdeI-LaCPR1(ΔN58)-XhoI構建成功,并在NdeI和XhoI酶切位點之間含有SEQ ID NO:2多核苷酸序列中175-2088位核苷酸。所獲得的重組質粒命名為pET28a-LaCPR1(ΔN58)。
實施例3、LaCPR1和LaCPR1(ΔN58)的表達、純化與酶活測定
(1)將重組質粒pET28a-LaCPR1利用熱擊法(42℃,90s)轉化進入大腸桿菌Rosseta(DE3)感受態細胞中,獲得重組大腸桿菌Rosseta(DE3)/pET28a-LaCPR1。挑取單克隆過夜培養,再將菌液按100倍稀釋于含卡那霉素(終濃度為25μg/mL)的LB培養基中培養。待菌液生長至600nm波長下吸光度為0.6-0.8時,加入誘導劑異丙基-β-D-硫代半乳糖苷(IPTG)(終濃度為0.1mmol/L)進行誘導培養,培養溫度為25℃。取誘導過的菌液40ml,離心收集菌體(4000rpm,10min,4℃),丟棄上清后用冰冷的1×PBS緩沖液洗滌菌體2遍,重懸于30ml 1×PBS緩沖液中。取1ml重懸菌液備用,剩余菌液進行超聲破碎細胞,然后高速低溫冷凍離心(12000g,30min,4℃)分離可溶部分和不可溶部分。取菌液、可溶部分和不可溶部分各400μl,分別加100μl 5×SDS Loading Buffer,短暫漩渦震蕩后,于沸水中煮5min,10000rpm離心10min,各取10μl樣品進行聚丙烯酰胺凝膠電泳(SDS-PAGE)檢測重組蛋白,結果見圖4,表達蛋白的分子量約為77KDa。取細菌破碎液上樣到鎳柱(購自南京金斯瑞生物科技有限公司),待濾液全部過濾完后,用Wash Buffer(20mM Tris-HCl,500mM NaCl,50mM imidazole,pH7.9)沖洗2次。然后用Elution Buffer(20mM Tris-HCl,500mM NaCl,250mM imidazole,pH7.9)洗脫。用Vivaspin濃縮洗脫液,并經PD-10脫鹽柱(GE Healthcare)脫鹽后,溶解于含10%甘油的Tris-HCl緩沖液中(50mM,pH8.0),進行聚丙烯酰胺凝膠電泳(SDS-PAGE)檢測重組蛋白,并采用Bradford方法(Bradford et al.,1976)測定蛋白濃度,然后分裝保存于-80℃備用。取純化的LaCPR1蛋白液利用Guengerich等的方法(Guengerich et al.,Nature Protocol,2009)測定酶活,結果如圖5,LaCPR1主要傳遞電子供體還原型煙酰胺腺嘌呤二核苷酸磷酸(NADPH)(還原力)中的電子。
(2)將重組質粒pET28a-LaCPR1(ΔN58)利用熱擊法(42℃,90s)轉化進入大腸桿菌Rosseta(DE3)感受態細胞中,獲得重組大腸桿菌Rosseta(DE3)/pET28a-LaCPR1(ΔN58)。挑取單克隆過夜培養,再將菌液按100倍稀釋于含卡那霉素(終濃度為25μg/mL)的LB培養基中培養。待菌液生長至600nm波長下吸光度為0.6-0.8時,加入誘導劑異丙基-β-D-硫代半乳糖苷(IPTG)(終濃度為0.1mmol/L)進行誘導培養,培養溫度為25℃。取誘導過的菌液40ml,離心收集菌體(4000rpm,10min,4℃),丟棄上清后用冰冷的1×PBS緩沖液洗滌菌體2遍,重懸于30ml 1×PBS緩沖液中。取1ml重懸菌液備用,剩余菌液進行超聲破碎細胞,然后高速低溫冷凍離心(12000g,30min,4℃)分離可溶部分和不可溶部分。取菌液、可溶部分和不可溶部分各400μl,分別加100μl 5×SDS Loading Buffer,短暫漩渦震蕩后,于沸水中煮5min,10000rpm離心10min,各取10μl樣品進行聚丙烯酰胺凝膠電泳(SDS-PAGE)檢測重組蛋白,結果見圖6,表達蛋白的分子量約為73KDa,其主要分布在大腸桿菌細胞破碎液的可溶性部分。取細菌破碎液上樣到鎳柱(購自南京金斯瑞生物科技有限公司),待濾液全部過濾完后,用Wash Buffer(20mM Tris-HCl,500mM NaCl,50mM imidazole,pH7.9)沖洗2次。然后用Elution Buffer(20mM Tris-HCl,500mM NaCl,250mM imidazole,pH7.9)洗脫。用Vivaspin濃縮洗脫液,并經PD-10脫鹽柱(GE Healthcare)脫鹽后,溶解于含10%甘油的Tris-HCl緩沖液中(50mM,pH8.0),進行聚丙烯酰胺凝膠電泳(SDS-PAGE)檢測重組蛋白,并采用Bradford方法(Bradford et al.,1976)測定蛋白濃度,然后分裝保存于-80℃備用。取純化的LaCPR1(ΔN58)蛋白液利用Guengerich等的方法(Guengerich et al.,Nature Protocol,2009)測定酶活,結果如圖7,LaCPR1(ΔN58)的酶活是LaCPR1的2.23倍。
實施例4、LaCPR1和CYP73A重組表達載體的構建
(1)合成分別具有序列表中SEQ ID NO:8和SEQ ID NO:9核苷酸序列的兩條引物,以忽地笑的cDNA為模板進行PCR擴增CYP73A編碼基因。PCR擴增程序同實施例1。PCR產物經瓊脂糖凝膠電泳分離、回收。回收獲得的DNA片段兩端分別帶有25bp與pET28a-LaCPR1載體XhoI酶切位點兩側同源的序列。利用XhoI限制性內切酶(購自寶生物工程(大連)有限公司(TaKaRa))將pET28a-LaCPR1載體線性化。利用One-step cloning試劑盒(購自南京諾唯贊生物科技有限公司)按照產品說明書將CYP73A編碼基因導入pET28a-LaCPR1載體的XhoI酶切位點中,利用合成的具有序列表中SEQ ID NO:10核苷酸序列的引物與通用測序引物T7ter進行菌落PCR驗證,獲得重組表達質粒pET28a-LaCPR1-CYP73A,結果如圖8。進一步測序證實重組質粒中LaCPR1和CYP73A進行了基因的融合表達。
(2)合成分別具有序列表中SEQ ID NO:8和SEQ ID NO:9核苷酸序列的兩條引物,以忽地笑的cDNA為模板進行PCR擴增CYP73A編碼基因。PCR擴增程序同實施例1。PCR產物經瓊脂糖凝膠電泳分離、回收。回收獲得的DNA片段兩端分別帶有25bp與pET28a-LaCPR1(ΔN58)載體XhoI酶切位點兩側同源的序列。利用XhoI限制性內切酶(購自寶生物工程(大連)有限公司(TaKaRa))將pET28a-LaCPR1(ΔN58)載體線性化。利用One-step cloning試劑盒(購自南京諾唯贊生物科技有限公司)按照產品說明書將CYP73A編碼基因導入pET28a-LaCPR1(ΔN58)載體的XhoI酶切位點中,利用合成的具有序列表中SEQ ID NO:10核苷酸序列的引物與通用測序引物T7ter進行菌落PCR驗證,獲得重組表達質粒pET28a-LaCPR1(ΔN58)-CYP73A,結果如圖9。進一步測序證實重組質粒中LaCPR1(ΔN58)和CYP73A進行了基因的融合表達。
(3)合成分別具有序列表中SEQ ID NO:11和SEQ ID NO:12核苷酸序列的兩條引物,在合成的引物SEQ ID NO:11和SEQ ID NO:12的5’-端分別設置NdeI和XhoI兩個酶切位點及其保護堿基序列,以忽地笑的cDNA為模板進行PCR擴增CYP73A編碼基因。PCR擴增程序同實施例1。PCR擴增產物經瓊脂糖凝膠電泳檢測、分離、切膠回收后經NdeI和XhoI雙酶切,利用T4DNA連接酶(購自寶生物工程(大連)有限公司(TaKaRa))連接同樣經NdeI和XhoI雙酶切的pET28a載體(Novagen)中。連接產物轉化大腸桿菌(E.coli)DH5α(購自南京諾唯贊生物科技有限公司)感受態細胞,并涂布于添加卡那霉素(終濃度為25μg/mL)的LB平板上。利用通用測序引物對T7和T7ter進行菌落PCR驗證陽性轉化子,獲得重組表達質粒pET28a-CYP73A,菌落PCR產物的瓊脂糖電泳結果如圖10。
實施例5、LaCPR1和LaCPR1(ΔN58)協同CYP73A催化反式-肉桂酸合成對-香豆酸
(1)配制培養基。誘導培養基(1L):31g Na2HPO4,15g KH2PO4,2.5g NaCl,5.0g NH4Cl,0.24g MgSO4,0.01g CaCl2,10g Glucose,1mL微量元素母液。生物轉化培養基(1L):31g Na2HPO4,15g KH2PO4,2.5g NaCl,0.24g MgSO4,0.01g CaCl2,20g Glucose,1mL微量元素母液。微量元素母液配方為(1L):0.15mM(NH4)6Mo7O24,20.0mM H3BO3,1.5mM CoCl2,0.5mM CuSO4,4.0mM MnCl2,0.5mM ZnSO4,100mM HCl。
(2)將重組質粒pET28a-LaCPR1、pET28a-CYP73A、pET28a-LaCPR1-CYP73A和pET28a-LaCPR1(ΔN58)-CYP73A分別轉化進入大腸桿菌Rosseta(DE3)細胞,獲得重組菌株Rosseta(DE3)/pET28a-LaCPR1、Rosseta(DE3)/pET28a-CYP73A、Rosseta(DE3)/pET28a-LaCPR1-CYP73A和Rosseta(DE3)/pET28a-LaCPR1(ΔN58)-CYP73A,分別命名為菌株EcR-LaCPR1、EcR-CYP73A、EcR-LaCPR1-CYP73A和EcR-LaCPR1(ΔN58)-CYP73A。每個菌株各挑取單克隆接種添加卡那霉素(終濃度為25μg/ml)的LB培養基于37℃、200rpm過夜培養。
(3)將過夜培養菌液按100倍稀釋于含卡那霉素(終濃度為25μg/mL)的50mL誘導培養基中培養。待菌液生長至600nm波長下吸光度為0.6-0.8時,加入誘導劑異丙基-β-D-硫代半乳糖苷(IPTG)(終濃度為0.1mmol/L)進行誘導培養,誘導時間為12h,誘導溫度為25℃。
(4)收集誘導12h的細菌培養液于8000rpm、4℃離心5min,棄上清后用40mL生物轉化培養基洗滌菌體1次。菌體用40ml生物轉化培養基重懸后全部轉移到250ml無菌三角瓶中,加入反式-肉桂酸(購自生工生物工程(上海)股份有限公司)二甲基亞砜(DMSO)母液至終濃度為100μM,于25℃、250rpm震蕩培養60h。
(5)取生物轉化液1ml于4℃凍融,加入等體積的甲醇,振蕩混勻。室溫下,12000rpm,離心5min。取上清用0.22μm孔徑濾膜過濾,濾液上樣高效液相色譜儀(HPLC)進行分析。分析條件為:采用LC-20A高效液相色譜儀(島津,日本),InertSustain C18色譜柱(5μm,4.6mm×250mm),30℃柱溫,二極管陣列檢測器,278nm和314nm波長,10μl進樣量,1.5%(v/v)乙酸水溶液為流動相A,100%乙腈為流動相B,0.9mL/min流速,梯度洗脫。分析結果如圖11,結果表明:LaCPR1和CYP73A單獨存在時并不能夠催化底物反式-肉桂酸合成產物對-香豆酸;只有在LaCPR1或LaCPR1(ΔN58)的存在下,CYP73A才能夠催化底物反式-肉桂酸合成產物對-香豆酸。可以理解為,LaCPR1和LaCPR1(ΔN58)具有細胞色素P450還原酶活性,可用于傳遞電子給細胞色素P450酶并協助細胞色素P450酶對底物的轉化和產物的合成。
實施例6、重組大腸桿菌Ec31884-LaCPR1(ΔN58)-CYP73A-PAL的構建
(1)合成分別具有序列表中SEQ ID NO:13和SEQ ID NO:14核苷酸序列的兩條引物,以忽地笑的cDNA為模板進行PCR擴增苯丙氨酸氨基裂解酶(PAL)編碼基因。PCR擴增程序為:95℃ 5min;94℃ 45s,56℃ 45s,72℃3.5min,共30個循環;72℃ 10min,降至10℃。PCR產物經瓊脂糖凝膠電泳分離、回收。回收獲得的DNA片段兩端分別帶有25bp與pET28a-LaCPR1(ΔN58)-CYP73A載體XhoI酶切位點兩端同源的序列,并且PAL基因起始密碼子上游12bp處添加了核糖體結合位點(rbs)序列(AGGAG)。利用XhoI限制性內切酶(購自寶生物工程(大連)有限公司(TaKaRa))將pET28a-LaCPR1(ΔN58)-CYP73A載體線性化。利用One-step cloning試劑盒(購自南京諾唯贊生物科技有限公司)按照產品說明書將CYP73A編碼基因導入pET28a-LaCPR1(ΔN58)-CYP73A載體的XhoI酶切位點中,利用合成的具有序列表中SEQ ID NO:13核苷酸序列的引物與通用測序引物T7ter進行菌落PCR驗證,獲得重組表達質粒pET28a-LaCPR1(ΔN58)-CYP73A-rbsPAL,結果如圖12。進一步測序證實重組質粒中PAL與LaCPR1(ΔN58)-CYP73A形成了多順反子基因結構。
(2)利用λDE3溶原化試劑盒(購自Novagen)按照試劑盒說明書操作,將λDE3原噬菌體插入到大腸桿菌苯丙氨酸生產菌株ATCC31884(購自ATCC)基因組中,獲得溶原化的宿主菌株大腸桿菌ATCC31884(DE3)。
(3)將重組質粒pET28a-LaCPR1(ΔN58)-CYP73A-rbsPAL轉化進入大腸桿菌ATCC31884(DE3)細胞,獲得重組菌株ATCC31884(DE3)/pET28a-LaCPR1(ΔN58)-CYP73A-rbsPAL,命名為菌株Ec31884-LaCPR1(ΔN58)-CYP73A-PAL。
實施例7、重組大腸桿菌Ec31884-LaCPR1(ΔN58)-CYP73A-PAL發酵合成對-香豆酸
(1)配制發酵培養基(1L):31g Na2HPO4,15g KH2PO4,2.5g NaCl,5.0g NH4Cl,0.24g MgSO4,0.01g CaCl2,30g Glucose,1mL微量元素母液。微量元素母液配方同實施例5。
(2)挑取菌株Ec31884-LaCPR1(ΔN58)-CYP73A-PAL單克隆接種添加卡那霉素(終濃度為25μg/ml)的LB培養基于37℃、200rpm過夜培養。
(3)將過夜培養菌液按100倍稀釋于含卡那霉素(終濃度為25μg/mL)的50mL發酵培養基中培養。待菌液生長至600nm波長下吸光度為0.6-0.8時,加入誘導劑異丙基-β-D-硫代半乳糖苷(IPTG)(終濃度為0.1mmol/L)進行誘導。培養溫度轉變為25℃,發酵時間為72h。
(4)取發酵液1ml于4℃凍融,加入等體積的甲醇,振蕩混勻。室溫下,12000rpm,離心5min。取上清用0.22μm孔徑濾膜過濾,濾液上樣高效液相色譜儀(HPLC)進行分析。分析條件同實施例5。分析結果如圖13,結果表明:所構建的大腸桿菌菌株能夠合成對-香豆酸,產物的量為400mg/L。可以理解為,基于能夠合成CYP450酶的底物及其上游代謝產物的微生物菌株,LaCPR1(ΔN58)具有細胞色素P450還原酶活性,可用于傳遞電子給細胞色素P450酶并協助CYP450酶對底物的轉化和產物的合成。
本發明提及的所有參考文獻都在本申請中引用作為參考,就如同每一篇文獻被單獨引用作為參考那樣。此外應理解,在閱讀了本發明的上述內容之后,本領域技術人員可以對本發明作各種改動或修飾,這些等價形式同樣落于本申請所附權利要求書所限定的范圍。
- 還沒有人留言評論。精彩留言會獲得點贊!
- 修飾細胞色素p450酶的編碼核酸和其應用方法
- 一種改進細胞色素p450酶家族體外表達的方法
- 含有(e)-7-[4-(4-氟苯基)-6-異丙基-2-[甲基(甲基磺酰基)氨基]嘧啶-5基 ...的制作方法
- 含三氮雜螺[5.5]十一烷衍生物與細胞色素p450同工酶3a4抑制劑和/或p-糖蛋白抑制劑 ...的制作方法
- 細胞色素p450 bm-3 d168s變體酶以及應用其制備靛玉紅的方法
- 細胞色素p450 bm-3 d168r變體酶以及應用其制備靛玉紅的方法
- 用于檢測人細胞色素p450相關的基因多態位點的引物系統及其用途的制作方法
- 人細胞色素p450 3a5酶及nadph-細胞色素p450氧化還原酶共表達體系的制作方法
- 一種檢測黃鱔芳香化酶p450基因等位基因甲基化狀態的方法
- 四翅濱藜細胞色素p450基因克隆及其應用的制作方法