一種伊潘立酮超細粉體的制備方法與流程
本發明專利涉及化學醫藥領域,特別是涉及一種伊潘立酮原料藥超細粉的制備方法。
背景技術:
伊潘立酮(iloperidone),3-甲氧基-4-[3-[4-(6-氟-1,2-苯并異噁唑-3-基)哌啶基]丙氧基]苯乙酮,其結構如式I所示。伊潘立酮片(商品名:Fanapt)由美國Vanda公司開發,并于2009年5月6日獲得FDA批準在美國上市,分為7種規格:1,2,4,6,8,10,12mg,輔料為乳糖,微晶纖維素,羥丙基甲基纖維素,交聯聚維酮,硬脂酸鎂,微粉硅膠和純化水(制備過程中出去)。
作為一種非典型抗精神病藥物,伊潘立酮對減輕精神分裂癥患者的陽性癥狀和陰性癥狀均有明顯效果。與目前使用的抗精神藥物比較,伊潘立酮的不良反應較少,患者體質量增加幅度較小,不會誘導患者發生糖尿病,患者錐體外系癥狀也較少。
伊潘立酮,水中的溶解度0.012mg/ml,為難溶性藥物,制備成片劑,藥物的溶出度會受到溶解度的影響。對于此類難溶性物料,藥物溶出成為限制其吸收及生物利用度的關鍵因素。根據Noyes-Whimey方程,減少藥物顆粒粒度,增加其比表面和潤濕性,從而達到顆粒的快速溶解的目的。因此,原料藥的微粉化成為解決難溶性藥物溶出度問題的重要途徑之一。
其中原料藥的微粉化包括重結晶、機械粉碎、振動磨粉碎及氣流粉碎等。
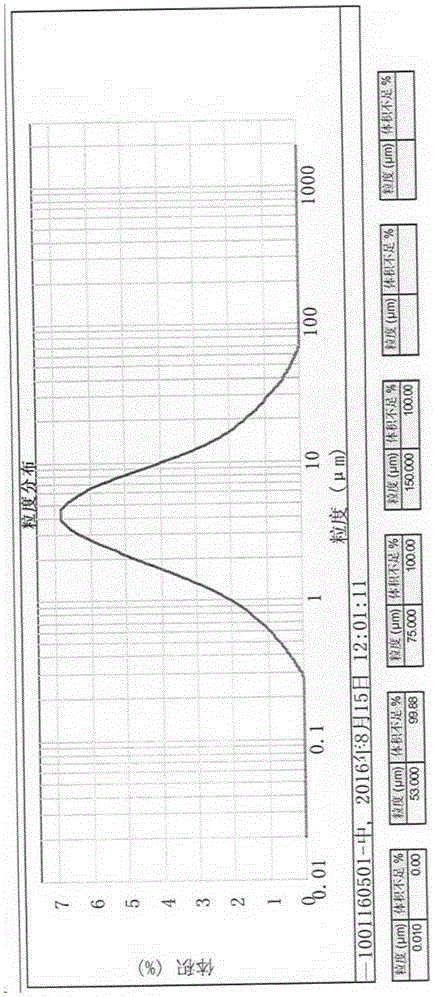
官方報道伊潘立酮為單一晶型,我們也通過大量單一溶劑進行重結晶的實驗研究進行了證實,該產品無多晶型現象且易于結晶,其基本的粒度均在50μm以上,自制放大產品的粒度接近150μm,詳見附圖1。后通過機械粉碎及氣流粉碎等處理,晶型無明顯變化,但均存在分散、靜電吸附等現象,活性成分損失大,超過近10%以上的損耗;同時在物理粉碎特別是氣流粉碎時,由于高溫放熱,導致伊潘立酮的雜質特別是氧化降解雜質增加,從而使制劑產品無法達到藥用標準。
另外,CN104324008A報道了采用二元溶劑(易溶溶劑及反溶劑)進行沉淀微粉化的方法制備超細粉伊潘立酮,該工藝操作簡單,但均采用了大量有機溶劑,該方法不利于環保。
我們遵循綠色化學的原則,采用水為主溶劑,通過酸堿中和以及調節pH析晶的策略(利用伊潘立酮在水中的過飽和度極低的特點進行快速析晶)進行了大量的實驗發現,大多數伊潘立酮的鹽(比如鹽酸鹽、硫酸鹽、硝酸鹽、草酸鹽、酒石酸鹽、枸櫞酸鹽、富馬酸鹽等)在水中溶解度均不很好,只有伊潘立酮的醋酸鹽水中溶解性好,而且可以通過調節pH達到控制超細粉粒粒度的產品。
但是,采用純水單一溶媒體系,產品析出過快過細,易于形成部分細微顆粒結塊及團的問題,干燥后仍需通過粉碎過篩后才能用于制劑產品的制備。
另外采用常用的無機堿如碳酸氫鈉、碳酸鈉等時,亦產生氣體,不利于生產操作,故選擇氫氧化鈉、氫氧化鉀等無機堿。綜合考慮綠色化學的基本原則,本發明不增加引入相關有機堿,故未做相關深入研究。
后續通過大量工藝優化的實驗,我們驚奇地發現,通過補充少量的有機溶劑,充分利用醋酸鹽析晶的合適速率,比較穩定可靠地直接獲得粒度分布比較均一的超細粉伊潘立酮原料藥,該產品可直接用于制劑的制備。
技術實現要素:
本發明的目的在于提供一種伊潘立酮原料藥超細粉的制備方法,該工藝綠色環保,操作簡單安全,工藝穩定,損耗少且成本低,易于產業化生產,特別適合直接用于伊潘立酮制劑的開發使用,亦可大大降低其氧化降解雜質的控制風險。
本發明的詳細技術方案概述如下:
一種伊潘立酮原料超細粉體的制備方法,包括以下步驟:
1)按比例將0.8-1.2g伊潘立酮懸浮于10~100ml水與的有機溶劑的混合溶劑中;
2)加入醋酸使伊潘立酮溶解;
3)在15~30℃,攪拌下滴加堿的水溶液,調節溶液pH=4-4.5;
4)析出晶體,放置;
5)在40~100℃干燥至恒重直接得到伊潘立酮超細粉體。
本發明所述的制備方法,所述步驟(1)中的有機溶劑為甲醇、乙醇、丙酮、丁酮、乙腈及他們的混合物,其體積和水體積的比例為1-4:20。
本發明所述的制備方法,所述步驟(2)加入醋酸的的摩爾量是伊潘立酮質量的1~5倍。
本發明所述的制備方法,步驟(2)中的堿選自氫氧化鈉、氫氧化鉀。
本發明所述的制備方法,步驟(4)中的放置時間,為30min~12h。
本發明所述的制備方法,步驟(5)中的粒度為D(90)≤20μm。
本發明是將伊潘立酮懸浮于水與一定比例的有機溶劑混合體系中,加入一定量的醋酸溶解后,然后在一定溫度下,充分攪拌下逐漸滴加一定量堿水溶液,析出晶體,再經過一定時間的陳化后,經過濾,干燥后無需粉碎直接得到顆粒分布相對均勻的伊潘立酮超細粉體產品,粒度均在20μm以下。本發明的方法,工藝綠色環保,操作簡單安全,工藝穩定,損耗少且成本低,易于產業化放大生產,特別適合直接用于伊潘立酮制劑的開發使用,亦可大大降低其氧化降解雜質的控制風險。
本發明的制備方法是通過篩選獲得的,本發明的篩選過程如下:
1)首先,伊潘立酮的酸堿等化學穩定性很好且晶型穩定,本文初步選擇通過酸堿反應造粒的方式制備其超細粉API。
2)伊潘立酮在水中的溶解度0.012mg/ml水溶性,采用水為主要溶媒是最佳選擇。
3)自制了大量伊潘立酮的鹽,比如醋酸鹽、鹽酸鹽、硫酸鹽、硝酸鹽、草酸鹽、酒石酸鹽、枸櫞酸鹽、富馬酸鹽等,其中,伊潘立酮的鹽酸鹽、硫酸鹽、硝酸鹽等在水中極微溶解,其草酸鹽、酒石酸鹽、枸櫞酸鹽、富馬酸鹽等在水中微溶,僅醋酸鹽在水中易溶,故選擇其作為合適的酸根進行后續開發。
4)對不同堿的篩選上,常見有機堿比如三乙胺或氨水等不易產生沉淀,且新增有機試劑不符合綠色化學的基本原則。無機堿比如碳酸鈉、碳酸鉀等則易于生成氣體,不利于工藝操作的穩定性及安全性。采用氫氧化鈉、氫氧化鉀等一元強堿,產品質量穩定性及工藝操作性均符合要求。
5)當采用單一純水體系進行酸堿反應造粒的時候,對滴加速度的控制要求太高,常因為顆粒太小而聚團結塊,通過離心及干燥后,仍需要粉碎處理后用于制劑使用。這里我們嘗試幾種常見的與水混溶且不與伊潘立酮溶解的溶劑比如甲醇、乙醇、丙酮、丁酮、乙腈等,效果十分明顯。考慮到有機溶劑含量過大析晶緩慢,顆粒增大,故進行了相關研究。
和現有技術的對比實驗:
1)我們采用氣流進行粉碎,顆粒過細,且損失在15%左右,相關控制不當,其氧化雜質易于增加,不適合現有伊潘立酮的制劑工藝。
2)我們重復了CN104324008A的相關工藝,在合適的工藝參數范圍內,產品粒度等相關質量基本能達到我們的要求,但有機溶劑種類太多,粒度偏細且殘溶量偏大。
經過篩選以及和現有技術的對比實驗,本發明意外的發現,本發明的存在以下優點:
1)綠色環保,溶殘量極低
2)產品實際收率均在90%以上,無特殊設備要求,成本相對低
3)產品粒度不聚團,分布均勻,可直接用于制劑生產
4)操作簡單安全易控,有利于工業化生產
5)質量可靠,降解雜質及殘留溶劑等控制十分合理
附圖說明
圖1中試自制伊潘立酮原料藥的粒度分布圖
圖2實施例1中伊潘立酮原料藥的粒度分布圖
具體實施方式
粒度測量是通過馬爾文SM2000型激光粒度儀進行采集:稱取約120mg伊潘立酮原料藥,置50ml的量瓶中,加入3.0ml的5%卵磷脂溶液和30ml的伊潘立酮Isopar G飽和溶液,超聲6min。運行背景測量之后,取溶液加入樣品池中測量,顆粒折射率1.430,顆粒吸收率0.8,遮光度5~20%,攪拌速度1600rpm,背景和樣品測量時間均為10s。
通過下面的實施例可以對本發明進行進一步的描述,然而本發明的范圍并沒有局限于下述實施例。
本發明采用自制放大的伊潘立酮API為原料,粒度測試如圖1,粒徑D90=142.9μm。
實施例1
(1)按比例將100g(0.234mol)伊潘立酮懸浮于1500ml水與100ml乙醇混合體系中;
(2)加入的醋酸(1.2eq)溶解后;
(3)然后在15~30℃下,充分攪拌下逐漸滴加一定量堿的水溶液,調節pH=4左右;
(4)析出晶體,再經過一定時間的陳化后;
(5)在40~100℃干燥至恒重直接得到96.2g,收率為96.2%,D90=13.7μm。粒度測試如圖2所示。
實施例2
同實例1,僅將乙醇體積從100ml改為250ml,得到固體產品92.6g,收率92.6%,D90=15.2μm。
實施例3:
同實例1,僅有機溶劑換為丙酮,得到固體產品95.1g,收率95.1%,D90=11.2μm。
實施例4:
同實例1,僅有機溶劑換為乙腈,得到固體產品96.7g,收率96.7%,D90=9.2μm。
實施例5:
同實例1,僅將其中100ml中的50ml乙醇換為50ml乙腈,得到固體產品96.3g,收率96.3%,D90=10.8μm。
- 還沒有人留言評論。精彩留言會獲得點贊!