合成5,6,4'?三羥基黃酮?7?0?D?葡萄糖醛酸有關物質及其制備方法和應用與流程
本發明涉及藥物化學技術領域,尤其涉及一種合成5,6,4'-三羥基黃酮-7-0-d-葡萄糖醛酸有關物質及其制備方法和應用。
背景技術:
5,6,4'-三羥基黃酮-7-0-d-葡萄糖醛酸,又名燈盞花乙素、野黃芩苷,是從菊科飛蓬屬植物短亭飛蓬(eriger0nbreviscapus)的全草(又名燈盞細辛)中提取的黃酮類混合物的主要有效成分,分子式為c21h18012,分子量為462.37,其結構式如式ⅳ所示:
臨床研究表明5,6,4'-三羥基黃酮-7-0-d-葡萄糖醛酸對高血壓、腦出血、腦血栓形成,腦栓塞多發性神經炎,慢性蛛網膜炎及其后遺癥具有較好療效,并對風濕病、冠心病也有一定療效。此外大量的研究證實5,6,4'-三羥基黃酮-7-0-d-葡萄糖醛酸具有抗炎、抗氧化、蛋白激酶抑制劑作用,還具有對糖尿病、腎病、風濕類風濕等疾病的治療作用。因此,目前5,6,4'-三羥基黃酮-7-0-d-葡萄糖醛酸臨床上廣泛用于治療腦血栓、腦梗塞、多發性神經炎、慢性蛛網膜等腦血管意外所致癱瘓以及冠心病、心絞痛、治療痛風性關節炎等癥。
目前除了植物提取的合成5,6,4'-三羥基黃酮-7-0-d-葡萄糖醛酸,合成5,6,4'-三羥基黃酮-7-0-d-葡萄糖醛酸作為改良型創新原料藥,具有良好的市場前景。但是合成5,6,4'-三羥基黃酮-7-0-d-葡萄糖醛酸雜質譜不同于目前上市的植物提取5,6,4'-三羥基黃酮-7-0-d-葡萄糖醛酸,對于合成5,6,4'-三羥基黃酮-7-0-d-葡萄糖醛酸的有關物質目前尚無研究。雜質的存在將影響產品使用的安全性,按照新藥開發過程中雜質控制的相關指導原則的要求,應對樣品中雜質進行徹底研究,嚴格控制。
技術實現要素:
有鑒于此,本發明要解決的技術問題在于提供一種合成5,6,4'-三羥基黃酮-7-0-d-葡萄糖醛酸有關物質及其制備方法和應用。
本發明提供的合成5,6,4'-三羥基黃酮-7-0-d-葡萄糖醛酸有關物質或其鹽,結構如式(ⅰ)所示:
本發明所述的合成5,6,4'-三羥基黃酮-7-0-d-葡萄糖醛酸是指通過化學合成的方法制備的5,6,4'-三羥基黃酮-7-0-d-葡萄糖醛酸。具體制備方法參照昆藥集團申請號分別為cn201210114758.9,cn201210114894.8,pct/cn2012/086543的專利申請。
所述合成5,6,4’-三羥基黃酮-7-0-d-葡萄糖醛酸含量大于99%,其中含有微量的合成5,6,4'-三羥基黃酮-7-0-d-葡萄糖醛酸有關物質,含量低于0.5%。
本發明還提供了上述合成5,6,4'-三羥基黃酮-7-0-d-葡萄糖醛酸有關物質的制備方法,包括以下步驟:
1)式(ⅱ)所示化合物在催化劑的作用下,進行選擇性消除反應,生成式(ⅲ)所示化合物;所述催化劑為1,8-二氮雜二環十一碳-7-烯(dbu)、吡啶和4-二甲氨基吡啶(dmap)中的任意一種或幾種;
2)式(ⅲ)所示化合物進行水解反應,得到式(ⅰ)所示化合物;
其反應方程式如下:
本發明首先以1,8-二氮雜二環十一碳-7-烯(dbu)、吡啶和4-二甲氨基吡啶(dmap)中的任意一種或幾種作為催化劑,式(ⅱ)所示化合物在催化劑的作用下,進行選擇性消除反應,選擇性的脫去一分子水,生成式(ⅲ)所示化合物。上述催化劑有助于提高選擇性消除反應的速率和選擇性。
所述選擇性消除反應優選在溶劑中進行,在本發明的某些具體實施例中,所述溶劑選自二氯甲烷、三氯甲烷、乙腈、甲苯、四氫呋喃和1,4-二氧六環中的任意一種或幾種。
在本發明的某些具體實施例中,所述式(ⅱ)所示化合物與催化劑的摩爾比為1:(0.1~5),在某些實施例中為1:(0.2~3),在一些實施例中為1:1。
所述選擇性消除反應的溫度為10~30℃,反應時間為3~20h,在某些實施例中為4~6h,在一些實施例中為8h,在一些實施例中為10h。
優選的,所述步驟2)具體為:式(ⅲ)所示化合物在無機堿催化下進行水解反應,然后加入無機酸調節體系ph值至2~3,進行中和反應,得到式(ⅰ)所示化合物。
在本發明的某些具體實施例中,所述無機堿選自氫氧化鈉、氫氧化鋰、氫氧化鉀、碳酸鉀和碳酸鈉中的任意一種或幾種。
所述無機酸選自鹽酸、硫酸、乙酸、甲酸、硝酸和磷酸中的任意一種或幾種。
所述水解反應的溶劑選自丙酮、水、甲醇、乙醇、二氯甲烷、二甲基亞砜、四氫呋喃和二氧六環中的任意一種或幾種。
所述水解反應的溫度優選為-10~40℃,在某些實施例中為-8~0℃,在一些實施例中為-5℃;反應時間優選為1~10h,在某些實施例中為2~4h,在一些實施例中為3h,在一些實施例中為2h,在一些實施例中為4h。
所述式(ⅲ)所示化合物與無機堿的摩爾比優選為1:(0.1~50),在某些實施例中為1:(10~30),一些實施例中為1:25,一些實施例中1:15,一些實施例中1:20。
所述中和反應的溫度優選為-10℃~40℃,在一些實施例中為-5℃。
在本發明的某些具體實施例中,可以進一步采用常規方法如重結晶進行提純,得到符合要求純度的雜質對照品。
本發明通過hplc法、uv吸收法、核磁共振、ms譜分析對上述合成5,6,4'-三羥基黃酮-7-0-d-葡萄糖醛酸有關物質進行了結構驗證。
本發明所述5,6,4’-三羥基黃酮-7-0-d-葡萄糖醛酸有關物質的結構確證包括以下步驟:
步驟1、hplc法證實制備所得5,6,4’-三羥基黃酮-7-0-d-葡萄糖醛酸有關物質b與昆藥集團股份有限公司合成5,6,4’-三羥基黃酮-7-0-d-葡萄糖醛酸中有關物質(雜質)b的保留時間完全一致。
步驟2、uv譜圖證實制備所得5,6,4’-三羥基黃酮-7-0-d-葡萄糖醛酸有關物質b與昆藥集團股份有限公司合成5,6,4’-三羥基黃酮-7-0-d-葡萄糖醛酸中有關物質b特征峰完全一致。
步驟3、核磁共振氫譜、碳譜對制備所得5,6,4’-三羥基黃酮-7-0-d-葡萄糖醛酸有關物質b的化學結構進行了確證。
步驟4、經ms譜分析,證實昆藥集團股份有限公司合成5,6,4’-三羥基黃酮-7-0-d-葡萄糖醛酸中有關物質b,與制備所得5,6,4’-三羥基黃酮-7-0-d-葡萄糖醛酸有關物質b,具有相同的分子離子信號。
本發明所述結構確證方法步驟1中,色譜條件流動相和體積比為:流動相a:water/tfa=1000/1(v/v),流動相b:acet0nitrile/tfa=1000/1(v/v),樣品溶劑:acetonitrile/dmso=50/50(v/v)。
本發明所述結構確證方法步驟3和步驟4證實了5,6,4’-三羥基黃酮-7-0-d-葡萄糖醛酸雜質b的化學結構如式ⅰ所示。
本發明還提供了上述合成5,6,4'-三羥基黃酮-7-0-d-葡萄糖醛酸有關物質或其鹽或上述制備方法制備的合成5,6,4'-三羥基黃酮-7-0-d-葡萄糖醛酸有關物質,在合成5,6,4'-三羥基黃酮-7-0-d-葡萄糖醛酸或其制劑有關物質檢測時,作為對照品的應用。
具體的,可應用于合成5,6,4'-三羥基黃酮-7-0-d-葡萄糖醛酸或其制劑的雜質分析控制、藥理毒理研究等。
本發明以5,6,4’-三乙酰氧基黃酮-7-o-d-三乙酰氧基葡萄糖醛酸甲酯(結構式如式ⅱ所示)為原料,經選擇性消除和水解兩步反應,成功制備得到5,6,4’-三羥基黃酮-7-0-d-葡萄糖醛酸有關物質b,并進一步純化,最終獲得5,6,4’-三羥基黃酮-7-0-d-葡萄糖醛酸有關物質b單體,進行確證后得出該有關物質化學結構。該方法合成路線短、成本低、操作簡單,結果準確,產品純度高,反應收率高,產品易于純化,可用于合成5,6,4’-三羥基黃酮-7-0-d-葡萄糖醛酸樣品的雜質分析控制,有利于合成5,6,4’-三羥基黃酮-7-0-d-葡萄糖醛酸的質量控制,更好的保障了產品質量。
附圖說明
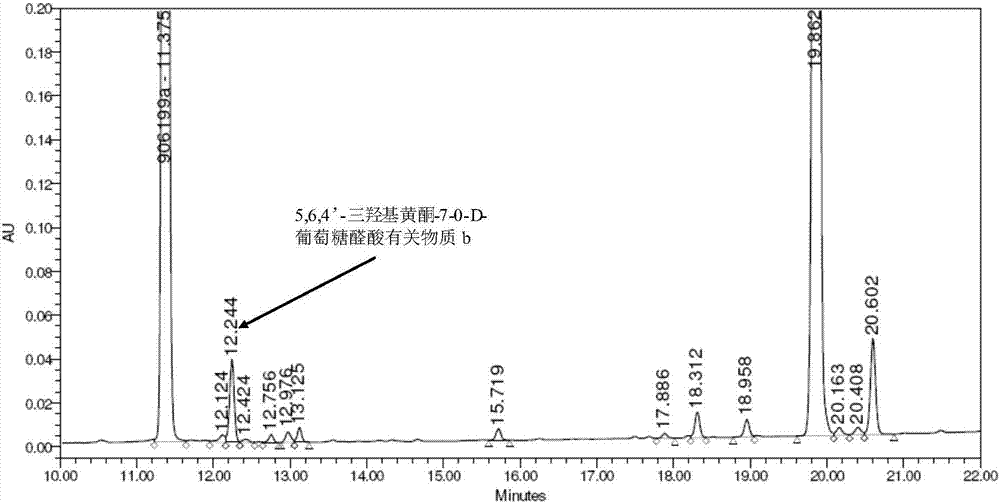
圖1是實施例1制備的5,6,4’-三羥基黃酮-7-0-d-葡萄糖醛酸有關物質b(批號:20140913)的hplc分析譜圖;
圖2是昆藥集團股份有限公司合成的5,6,4’-三羥基黃酮-7-0-d-葡萄糖醛酸(批號20140424-1)的hplc分析譜圖;
圖3是實施例1制備的5,6,4’-三羥基黃酮-7-0-d-葡萄糖醛酸有關物質b(批號20140913)以5%質量分數加入到5,6,4’-三羥基黃酮-7-0-d-葡萄糖醛酸(批號20140424-1)的hplc分析譜圖;
圖4是昆藥集團股份有限公司合成的5,6,4’-三羥基黃酮-7-0-d-葡萄糖醛酸(批號20140424-1)中5,6,4’-三羥基黃酮-7-0-d-葡萄糖醛酸有關物質b的uv譜圖;
圖5是實施例1制備的5,6,4’-三羥基黃酮-7-0-d-葡萄糖醛酸有關物質b(批號20140913)的uv譜圖;
圖6是實施例1制備的5,6,4’-三羥基黃酮-7-0-d-葡萄糖醛酸有關物質b(批號20140913)的hplc分析譜圖;
圖7是合成5,6,4’-三羥基黃酮-7-0-d-葡萄糖醛酸(批號20140424-1)+實施例1制備的5,6,4’-三羥基黃酮-7-0-d-葡萄糖醛酸有關物質b(20140913)的ms譜圖;
圖8是實施例1制備的5,6,4’-三羥基黃酮-7-0-d-葡萄糖醛酸雜質b(20140913)的ms譜圖;
圖9是合成5,6,4’-三羥基黃酮-7-0-d-葡萄糖醛酸(批號20140424-1)的ms譜圖。
具體實施方式
為了進一步說明本發明,下面結合實施例對本發明提供的合成5,6,4'-三羥基黃酮-7-0-d-葡萄糖醛酸有關物質(以下簡稱5,6,4'-三羥基黃酮-7-0-d-葡萄糖醛酸有關物質b)及其制備方法和應用進行詳細描述。
實施例1
(1)化合物ⅲ的制備
20g(27.4mmol)5,6,4’-三乙酰氧基黃酮-7-o-d–三乙酰氧基葡萄糖醛酸甲酯溶解于200ml甲苯中,然后攪拌下緩慢滴加入1,8-二氮雜二環十一碳-7-烯(dbu)4.1g(27mmol),30℃下攪拌反應約8小時反應完全。反應混合物攪拌下加入約500ml乙酸乙酯,用500ml水洗兩次。分出水層后的有機層中加入適量無水硫酸鈉干燥,減壓蒸干。硅膠柱層析并用石油醚:乙酸乙酯=5:1(v/v)的洗脫劑洗脫,得15g白色固體化合物ⅲ,色譜純度98.00%以上,收率為82%。
1h-nmr(400mhz,dms0),δ(ppm):7.95(2h,d,j=8.6hz),7.51(1h,s),7.32(2h,d,j=8.6hz),6.78(1h,s),6.15(1h,d,j=7.8hz),5.57(1h,d,j=10hz),5.38(1h,m),4.94(1h,m),3.67(3h,s),2.34(3h,s),2.31(3h,s),2.25(3h,s),2.02(3h,s),2.01(3h,s).
(2)5,6,4’-三羥基黃酮-7-0-d-葡萄糖醛酸有關物質b的制備
13.4g(20mmol)化合物ⅲ中加入200ml丙酮和200ml水的混合溶劑,在-5℃攪拌下緩慢滴加10%的naoh水溶液200ml,攪拌反應約三小時后,經tlc監測反應已完全。-5℃下緩緩向反應液中滴加2mol/l鹽酸水溶液中和反應液至ph為2~3,持續攪拌一小時并逐漸升至室溫,反應液中漸漸有黃色固體5,6,4’-三羥基黃酮-7-0-d-葡萄糖醛酸析出。室溫下靜置過夜,過濾,濾餅用適量水和丙酮洗,干燥后用乙醇重結晶得到8g5,6,4’-三羥基黃酮-7-0-d-葡萄糖醛酸有關物質b,批號記為20140913,收率為89%。hplc檢測,純度為98.88%,hplc檢測的色譜圖和統計結果見圖1和表1,表1是圖1的數據匯總。
1h-nmr(400mhz,dms0),δ(ppm):12.78(1h,s),10.51(1h,s),8.71(1h,s),7.93(2h,d,j=8.8hz),7.11(1h,s),6.94(2h,d,j=8.8hz),6.83(1h,s),5.99(1h,d,j=3.6hz),5.81(1h,d,j=5.6hz),5.74(1h,s),5.28(1h,s),4.14(1h,dd,j=4.0,4.5hz),3.84(1h,dd,j=4.5,6.0hz).
表1圖1的數據匯總
實施例2
(1)化合物ⅲ的制備
20g(27.4mmol)5,6,4’-三乙酰氧基黃酮-7-o-d–三乙酰氧基葡萄糖醛酸甲酯溶解于200ml二氯甲烷中,然后攪拌下緩慢加入4-二甲氨基吡啶(dmap)1.7g(13.7mmol,0.5eq),25℃下攪拌反應約10小時反應完全。反應混合物攪拌下加入約300ml乙酸乙酯,用500ml水洗兩次。分出水層后的有機層中加入適量無水硫酸鈉干燥,減壓蒸干。硅膠柱層析并用石油醚:乙酸乙酯=5:1(v/v)的洗脫劑洗脫,得14.3g白色固體化合物ⅲ,色譜純度98.00%以上,收率為78%。核磁鑒定結果與實施例1結果一致。
(2)5,6,4’-三羥基黃酮-7-0-d-葡萄糖醛酸有關物質b的制備
13.4g(20mmol)化合物ⅲ中加入200ml丙酮和200ml水的混合溶劑,在0℃攪拌下緩慢滴加10%的氫氧化鉀水溶液220ml,攪拌反應約四小時后,經tlc監測反應已完全。0℃下緩緩向反應液中滴加乙酸中和反應液至ph為2~3,持續攪拌一小時并逐漸升至室溫,反應液中漸漸有黃色固體5,6,4’-三羥基黃酮-7-0-d-葡萄糖醛酸有關物質b析出。室溫下靜置過夜,過濾,濾餅用適量水和丙酮洗,干燥后用乙醇重結晶得到7.4g5,6,4’-三羥基黃酮-7-0-d-葡萄糖醛酸有關物質b,收率為83%。hplc檢測,純度達98.00%以上,核磁鑒定結果與實施例1結果一致。
實施例3
(1)化合物ⅲ的制備
20g(27.4mmol)5,6,4’-三乙酰氧基黃酮-7-o-d–三乙酰氧基葡萄糖醛酸甲酯溶解于200ml四氫呋喃中,然后攪拌下滴加入吡啶6.5g(82.2mmol),25℃下攪拌反應約6小時反應完全。反應混合物攪拌下加入約200ml乙酸乙酯,用500水洗兩次。分出水層后的有機層中加入適量無水硫酸鈉干燥,減壓蒸干。硅膠柱層析并用石油醚:乙酸乙酯=5:1(v/v)的洗脫劑洗脫,得14.7g白色固體化合物ⅲ,色譜純度98.00%以上,收率為80%。核磁鑒定結果與實施例1結果一致。
(2)5,6,4’-三羥基黃酮-7-0-d-葡萄糖醛酸有關物質b的制備
13.4g(20mmol)化合物ⅲ中加入200ml丙酮和200ml水的混合溶劑,在5℃攪拌下緩慢滴加10%的氫氧化鋰水溶液190ml,攪拌反應約兩小時后,經tlc監測反應已完全。5℃下緩緩向反應液中滴加2mol/l硫酸水溶液中和反應液至ph為2~3,持續攪拌一小時并逐漸升至室溫,反應液中漸漸有黃色固體5,6,4’-三羥基黃酮-7-0-d-葡萄糖醛酸有關物質b析出。室溫下靜置過夜,過濾,濾餅用適量水和丙酮洗,干燥后用乙醇重結晶得到7.6g5,6,4’-三羥基黃酮-7-0-d-葡萄糖醛酸有關物質b,收率為85%。hplc檢測,純度達98.00%以上,核磁鑒定結果與實施例1結果一致。
實施例4結構確證
1、hplc確證合成制備所得化合物為合成5,6,4’-三羥基黃酮-7-0-d-葡萄糖醛酸中的5,6,4’-三羥基黃酮-7-0-d-葡萄糖醛酸有關物質b:
hplc色譜條件如下:
hplc系統:watersalliance2695四元泵
watersalliance2998pda檢測器
watersalliance2695自動進樣器
watersalliance2695柱溫箱
empower2網絡版工作站
色譜柱:agilentzorbaxsb-c8,5um,250x4.6mmpn:880975-906
流速:1.0ml/min.
檢測波長:uv254nm
進樣體積:10ul
柱溫:30℃
自動進樣器溫度:5℃
流動相a:water/tfa=1000/1(v/v)
流動相b:acetonitrile/tfa=1000/1(v/v)
樣品溶劑:acetonitrile/dms0=50/50
樣品濃度:1.0mg/ml
實施例1合成制備獲得的5,6,4’-三羥基黃酮-7-0-d-葡萄糖醛酸有關物質b(批號:20140913),與合成5,6,4’-三羥基黃酮-7-0-d-葡萄糖醛酸(批號20140424-1)中含有的雜質b的等同性驗證:
分別對合成制備所得5,6,4’-三羥基黃酮-7-0-d-葡萄糖醛酸有關物質b、合成5,6,4’-三羥基黃酮-7-0-d-葡萄糖醛酸進行hplc分析(譜圖如圖1、圖2所示),表2是圖2的數據匯總。再將約5%的5,6,4’-三羥基黃酮-7-0-d-葡萄糖醛酸有關物質b標準品,加入昆藥集團股份有限公司合成的5,6,4’-三羥基黃酮-7-0-d-葡萄糖醛酸樣品(批號20140424-1),進行hplc分析(譜圖如圖3所示),表3是圖3的數據匯總。
分析結果顯示,合成制備獲得的5,6,4’-三羥基黃酮-7-0-d-葡萄糖醛酸有關物質b標準品出峰位置與5,6,4’-三羥基黃酮-7-0-d-葡萄糖醛酸中本身含有的5,6,4’-三羥基黃酮-7-0-d-葡萄糖醛酸有關物質b完全吻合。
對二者的uv吸收進行檢測,譜圖如圖4、圖5所示,結果表明兩者的uv吸收圖譜完全一致。因此它們的等同性可以得到確認。
表2圖2的數據匯總
表3圖3的數據匯總
2、質譜和lc/ms解析
測試條件:
質譜:waterssqd
軟件:watersempower2網絡版
source:120℃desolvati0n:300℃capilary:3.5kv
lc-ms:
流動相a:超純水(milliporeultrapurewatersystem,resistivity:18.2mω.cm)+0.1%甲酸
pumpb:色譜純乙腈(merckkgaa64271,hypergradeforlc-ms)+0.1%甲酸
色譜柱:
柱溫:30℃
檢測波長:λ=254nm,230nmand218nm/pda
流速:1.0ml/min
譜圖見圖6、圖7、圖8和圖9。
由上述譜圖可知,對5,6,4’-三羥基黃酮-7-0-d-葡萄糖醛酸(批號20140424-1)中含有的雜質b,以及合成制備的5,6,4’-三羥基黃酮-7-0-d-葡萄糖醛酸雜質b(批號201409132)進行lc/ms分析,均顯示其分子量為444([m-h]-443)(見圖8和圖9)。由其分子量可以推測,雜質b應該是5,6,4’-三羥基黃酮-7-0-d-葡萄糖醛酸脫一分子水后的產物。并且,5,6,4’-三羥基黃酮-7-0-d-葡萄糖醛酸中本身含有的雜質b,與合成制備的雜質b,具有相同的分子離子信號。
3、5,6,4’-三羥基黃酮-7-0-d-葡萄糖醛酸雜質b的核磁共振譜圖解析
核磁共振氫譜:
儀器型號:brukeravanceiii400spectrometer(z115310,sn10032129)
溶劑:[d6]dmso(d,99.9%)+0.03%v/vtms,10*0.6mlampoule,cambridgeisotopelaboratories,inc.,lot12a-029
溫度:25℃
測試數據見表4。
表4核磁共振氫譜數據匯總
核磁共振13c譜:
儀器型號:brukeravanceiii400spectrometer(z115310,sn10032129)
溶劑:[d6]dmso(d,99.9%)+0.03%v/vtms,10*0.6mlampoule,cambridgeisotopelaboratories,inc.,lot12a-029
溫度:25℃
結果見表5。
表55,6,4’-三羥基黃酮-7-0-d-葡萄糖醛酸雜質b的核磁共振碳譜分析
由以上結果可知,ms一級譜給出了雜質準確的分子量信息:m/z442.7,與5,6,4’-三羥基黃酮-7-0-d-葡萄糖醛酸脫一分子水后所得的5,6,4’-三羥基黃酮-7-0-d-葡萄糖醛酸有關物質b分子量一致。hplc譜確證了制備所得5,6,4’-三羥基黃酮-7-0-d-葡萄糖醛酸有關物質b與合成5,6,4’-三羥基黃酮-7-0-d-葡萄糖醛酸中5,6,4’-三羥基黃酮-7-0-d-葡萄糖醛酸有關物質b的保留時間完全一致,且uv譜證實了二者的特征吸收峰完全一致。核磁共振氫譜和碳譜,證實了5,6,4’-三羥基黃酮-7-0-d-葡萄糖醛酸有關物質(雜質)b化學結構如式(ⅰ)所示:
由上述實施例可知,本發明證實了上述5,6,4’-三羥基黃酮-7-0-d-葡萄糖醛酸有關物質b的結構,并成功制備得到該化合物,且具有較高的純度,可以直接用于合成5,6,4’-三羥基黃酮-7-0-d-葡萄糖醛酸的雜質分析控制,以此基礎制訂了嚴格的雜質限度,為將來產品的安全使用提供了理論依據。
以上實施例的說明只是用于幫助理解本發明的方法及其核心思想。應當指出,對于本技術領域的普通技術人員來說,在不脫離本發明原理的前提下,還可以對本發明進行若干改進和修飾,這些改進和修飾也落入本發明權利要求的保護范圍內。
- 還沒有人留言評論。精彩留言會獲得點贊!
- 包含甲氧基黃酮的NOX抑制剤及NFκB抑制剤的制作方法與工藝
- 一種多甲氧基黃酮、組合物及其制劑用于預防或治療糖尿病的用途的制作方法與工藝
- 一種1?(2?羥基?3?甲氧苯基)乙酮的制備工藝的制作方法與工藝
- 一種多甲氧基黃酮在制備預防心血管炎癥藥物上的應用的制作方法與工藝
- 3’-羥基染料木黃酮用于制備抑制黑色素生成的組合物的用途的制作方法與工藝
- 合成5,6,4'?三羥基黃酮?7?0?D?葡萄糖醛酸有關物質及其制備方法和應用與流程
- 5?(4?羥基?3?甲氧基苯亞甲基)?2?(2?硝基苯亞胺基)噻唑烷酮及應用的制作方法與工藝
- 制備荷羥酮(甲基)丙烯酸酯的方法與流程
- 從甌柑果實中分離純化多甲氧基黃酮類化合物的方法與流程
- 一種多甲氧基黃酮在制備治療黑色素瘤的藥物上的應用的制造方法與工藝