一種烏苯美司的制備方法與流程
本發(fā)明涉及制藥領(lǐng)域,具體而言,涉及一種烏苯美司的制備方法。
背景技術(shù):
烏苯美司(iubenimex;nn),也稱為bestatin,化學(xué)式為n-[(2s,3r)-3-氨基-2-羥基-4-苯基丁酰基]-l-亮氨酸,是一種競(jìng)爭(zhēng)性的,可逆的蛋白酶抑制劑。它對(duì)精氨酰氨基肽酶(氨基肽酶b),白三烯a4水解酶(顯示環(huán)氧化物水解酶和氨基肽酶活性的鋅金屬蛋白酶),丙氨酰氨基肽酶(氨肽酶m/n),亮氨酰/胱氨酰氨基肽酶(催產(chǎn)素/血管加壓素酶)(白三烯d4水解酶)有抑制作用。正在研究用于治療急性骨髓性白血病。它源自橄欖鏈霉菌(streptomycesolivoreticuli)。已發(fā)現(xiàn)ubenimex抑制催產(chǎn)素,加壓素,腦啡肽和各種其它肽和化合物的酶促降解。
烏苯美司可增強(qiáng)免疫功能,用于抗癌化療、放療的輔助治療,老年性免疫功能缺陷等。可配合化療、放療及聯(lián)合應(yīng)用于白血病、多發(fā)性骨髓瘤、骨髓增生異常綜合癥及造血干細(xì)胞移植后,以及其它實(shí)體瘤患者。
現(xiàn)有技術(shù)中,烏苯美司在合成過(guò)程中大多采用四氫呋喃、三乙胺作為溶劑或者催化劑等,這些試劑本身不環(huán)保,后續(xù)處理負(fù)擔(dān)比較大,會(huì)產(chǎn)生大量的廢水廢氣,而且三乙胺這種有機(jī)堿容易殘留在產(chǎn)物中,造成有機(jī)溶劑殘留的問(wèn)題。
有鑒于此,特提出本發(fā)明。
技術(shù)實(shí)現(xiàn)要素:
本發(fā)明的第一目的在于提供一種烏苯美司的制備方法,該制備方法中摒棄了以往采用的有機(jī)堿、四氫呋喃、二氯甲烷、n、n二甲基甲酰胺等有機(jī)溶劑作為反應(yīng)溶劑、乙酸乙酯作為萃取溶劑。直接采用乙酸乙酯作為反應(yīng)和萃取溶劑,節(jié)約了成本同時(shí)避免了使用四氫呋喃、二氯甲烷、n、n二甲基甲酰胺所帶來(lái)的安全環(huán)保隱患,采用弱酸強(qiáng)堿無(wú)機(jī)鹽作為堿催化劑,降低了成本,同時(shí)避免三乙胺殘留溶劑對(duì)成品質(zhì)量的影響,整個(gè)制備方法過(guò)程中充分做到了避免使用有機(jī)溶劑,操作簡(jiǎn)便,綠色環(huán)保,在提高了烏苯美司產(chǎn)品質(zhì)量的同時(shí),收率高。
為了實(shí)現(xiàn)本發(fā)明的上述目的,特采用以下技術(shù)方案:
本發(fā)明提供了一種烏苯美司的制備方法,包括如下步驟:
(a)dcc(二環(huán)己基碳二亞胺)催化條件下,溶劑為乙酸乙酯,將l-亮氨酸芐酯對(duì)甲苯磺酸鹽、hobt(1-羥基苯并三氮唑)縮合得到活化酯溶液;
(b)將(2s,3r)-3-芐氧甲酰胺基-2-羥基-4-苯基丁酸、弱酸強(qiáng)堿無(wú)機(jī)鹽混合攪拌過(guò)程中,滴加所述活化酯溶液進(jìn)行反應(yīng)得到n-[(2s,3r)-3-芐氧甲酰胺基-2-羥基-4-苯基丁酰]-l-亮氨酸;
(c)n-[(2s,3r)-3-芐氧甲酰胺基-2-羥基-4-苯基丁酰]-l-亮氨酸在pd/c催化下,氫氣還原得到烏苯美司。
現(xiàn)有技術(shù)中,由于烏苯美司可增強(qiáng)免疫功能,用于抗癌化療、放療的輔助治療,老年性免疫功能缺陷等。可配合化療、放療及聯(lián)合應(yīng)用于白血病、多發(fā)性骨髓瘤、骨髓增生異常綜合癥及造血干細(xì)胞移植后,以及其它實(shí)體瘤患者,因此鑒于其廣泛的應(yīng)用,對(duì)其具體制備工藝的研究顯得非常具有實(shí)際意義。
但是,傳統(tǒng)的烏苯美司在合成過(guò)程中大多采用四氫呋喃、二氯甲烷、n、n二甲基甲酰胺、三乙胺作為溶劑或者催化劑等,這些試劑本身不環(huán)保,后續(xù)處理負(fù)擔(dān)比較大,會(huì)產(chǎn)生大量的廢水廢氣,而且三乙胺這種有機(jī)堿容易殘留在產(chǎn)物中,產(chǎn)生有機(jī)溶劑殘留的問(wèn)題。
本發(fā)明為了解決上述技術(shù)問(wèn)題,提供了一種烏苯美司的制備方法,該制備方法中,采用無(wú)機(jī)的弱酸強(qiáng)堿鹽代替?zhèn)鹘y(tǒng)的三乙胺這種有機(jī)堿,無(wú)機(jī)鹽最好選擇碳酸氫鈉或碳酸氫鉀,更優(yōu)選擇為碳酸氫鈉,由于溶于水時(shí)呈現(xiàn)弱堿性從而促進(jìn)反應(yīng)的順利進(jìn)行,但是需要注意的是,這里一定不要選擇堿性過(guò)強(qiáng)的鹽,因?yàn)槿绻麎A性過(guò)強(qiáng),l-亮氨酸芐酯對(duì)甲苯磺酸鹽的芐酯會(huì)脫落,從而影響到反應(yīng)的順利進(jìn)行,因此在進(jìn)行選擇時(shí)要選擇堿性比較適宜的鹽。
另外,本發(fā)明在溶劑選擇上選擇的為乙酸乙酯,改進(jìn)了傳統(tǒng)需要同時(shí)采用四氫呋喃、二氯甲烷、n、n二甲基甲酰胺中一種為反應(yīng)溶劑、乙酸乙酯為萃取溶劑。避免兩種溶劑同時(shí)使用。節(jié)約了成本的同時(shí)避免了使用四氫呋喃、二氯甲烷、n、n二甲基甲酰胺等所帶來(lái)的安全隱患。傳統(tǒng)工藝之所以需要采用四氫呋喃、二氯甲烷、n、n二甲基甲酰胺,是因?yàn)樗臍溥秽⒍燃淄椤、n二甲基甲酰胺本身溶解性好,原料(2s,3r)-3-芐氧甲酰胺基-2-羥基-4-苯基丁酸,還有hobt(1-羥基苯并三氮唑)能夠發(fā)生反應(yīng)的關(guān)鍵在于,其具有良好溶解性的介質(zhì)中充分分散以促進(jìn)反應(yīng)的正常進(jìn)行,但是本發(fā)明利用了dcc催化以先形成活化酯溶液,做成活化酯后采用乙酸乙酯為溶劑,其溶解性已經(jīng)可以滿足工藝要求,不需要采用四氫呋喃、二氯甲烷、n、n二甲基甲酰胺即保證反應(yīng)的順利進(jìn)行,并且通過(guò)采用弱酸強(qiáng)堿無(wú)機(jī)鹽也一部分解決了溶解性的問(wèn)題,提高了溶解性,可見(jiàn)本發(fā)明采用的弱酸強(qiáng)堿無(wú)機(jī)鹽不僅起到了催化作用,也間接起到了作為溶解介質(zhì)的作用。
本發(fā)明烏苯美司的制備工藝的總反應(yīng)方程式為:
步驟(a)中,優(yōu)選地,先添加l-亮氨酸芐酯對(duì)甲苯磺酸鹽、hobt,乙酸乙酯,并將溫度控制在0-5℃之間,然后添加含有dcc的乙酸乙酯溶液,將溫度升高至15-20℃之間,反應(yīng)4-5h,一般在后續(xù)真正發(fā)生反應(yīng)的時(shí)候?qū)囟壬咭员WC反應(yīng)的正常進(jìn)行,前期溫度在0-5℃之間是為了防止溫度太高副反應(yīng)的發(fā)生。
優(yōu)選地,含有dcc的乙酸乙酯溶液按照滴加的方式進(jìn)行,通過(guò)逐漸滴加的方式可以保證反應(yīng)平穩(wěn)的進(jìn)行,以控制反應(yīng)進(jìn)度,避免劇烈反應(yīng)。
步驟(b)中,將碳酸氫鈉最好配制成10-20wt%的碳酸氫鈉溶液,因?yàn)樘妓釟溻c通過(guò)配制成溶液,可以提高反應(yīng)原料的分散性,提高反應(yīng)的催化性能。
另外,添加碳酸氫鈉溶液后,溫度一般控制在10-15℃之間,并通過(guò)快速攪拌的方式以加快各原料之間的互溶。
優(yōu)選地,滴加活化酯溶液后,15-20℃之間反應(yīng)4-5h,得到目的產(chǎn)物,但是還是需要后續(xù)分離的步驟以純化目的產(chǎn)物,一般采用tlc來(lái)跟蹤反應(yīng)終點(diǎn)(乙酸乙酯:石油醚=2:1、紫外254nm)。
優(yōu)選地,反應(yīng)后將產(chǎn)物n-[(2s,3r)-3-芐氧甲酰胺基-2-羥基-4-苯基丁酰]-l-亮氨酸進(jìn)行一系列的分離步驟得到純化后的產(chǎn)物,具體步驟最好按照如下操作進(jìn)行:過(guò)濾分離有機(jī)相后,洗滌、干燥以及濃縮操作,干燥后得到濃縮物,向濃縮物中加入石油醚攪拌2-3h,過(guò)濾,干燥以濃縮,石油醚的添加也是為了將目的產(chǎn)物更好的分散在有機(jī)相里面。
優(yōu)選地,向濃縮物中加入石油醚攪拌2-3h,過(guò)濾后,40-45℃的條件下干燥,經(jīng)過(guò)干燥后得到分離純化后的產(chǎn)物,經(jīng)過(guò)測(cè)定該步驟的收率可以達(dá)到94%以上。
步驟(c)中,溶劑采用的為冰醋酸,反應(yīng)壓力控制在1.5-2mpa之間,反應(yīng)溫度控制在20-30℃之間。該步驟通過(guò)將n-[(2s,3r)-3-芐氧甲酰胺基-2-羥基-4-苯基丁酰]-l-亮氨酸催化還原,pd/c、氫氣的條件下脫掉cbz的保護(hù)基,得到最終的目的產(chǎn)物烏苯美司,但是此時(shí)得到的烏苯美司為粗品,后續(xù)通過(guò)酸溶堿沉的方法結(jié)晶以獲得成品。因此最好將反應(yīng)得到的烏苯美司粗品進(jìn)行過(guò)濾、分離以進(jìn)一步純化。
最后,后續(xù)的后處理步驟最好按照如下步驟進(jìn)行:將烏苯美司粗品在70-80℃、壓力-0.05~-0.1mpa之間進(jìn)行濃縮后,添加丙酮攪拌后,過(guò)濾,添加氨水析晶后,過(guò)濾即可。
優(yōu)選地,添加氨水后,調(diào)節(jié)溶液的ph值在5-6之間,0-5℃析晶4-5h。
還有優(yōu)選地,析晶后過(guò)濾加入丙酮攪拌2-3h,40-50℃之間干燥得到烏苯美司成品。
現(xiàn)有技術(shù)的一鍋法在合成n-[(2s,3r)-3-芐氧甲酰胺基-2-羥基-4-苯基丁酰]-l-亮氨酸的過(guò)程中使用的四氫呋喃、乙酸乙酯兩種溶劑,本發(fā)明的工藝僅使用乙酸乙酯作為溶劑,節(jié)約了成本的同時(shí)避免了使用四氫呋喃所帶來(lái)的安全隱患。還有,一鍋法在合成n-[(2s,3r)-3-芐氧甲酰胺基-2-羥基-4-苯基丁酰]-l-亮氨酸的過(guò)程中使用的是三乙胺為堿催化劑,本發(fā)明采用碳酸氫鈉為堿催化劑,節(jié)約成本的同時(shí),避免三乙胺殘留溶劑對(duì)成品質(zhì)量的影響。另外,一鍋法在合成n-[(2s,3r)-3-芐氧甲酰胺基-2-羥基-4-苯基丁酰]-l-亮氨酸的過(guò)程中需要進(jìn)行濃縮四氫呋喃,而本發(fā)明的工藝反應(yīng)結(jié)束直接萃取處理,更易操作,并不需要對(duì)四氫呋喃進(jìn)行單獨(dú)處理,省時(shí)省力,也比較環(huán)保。最后,一鍋法合成的n-[(2s,3r)-3-芐氧甲酰胺基-2-羥基-4-苯基丁酰]-l-亮氨酸收率無(wú)本發(fā)明的工藝高。
與現(xiàn)有技術(shù)相比,本發(fā)明的有益效果為:
(1)本發(fā)明的烏苯美司的制備方法,摒棄了以往采用的有機(jī)堿、四氫呋喃等有機(jī)溶劑的添加,有機(jī)溶劑僅僅采用乙酸乙酯,節(jié)約了成本同時(shí)避免了使用四氫呋喃所帶來(lái)的安全隱患,采用弱酸強(qiáng)堿無(wú)機(jī)鹽作為堿催化劑,節(jié)約成本的同時(shí),避免三乙胺殘留溶劑對(duì)成品質(zhì)量的影響,整個(gè)制備方法過(guò)程中避免使用有機(jī)溶劑,操作過(guò)程簡(jiǎn)便,容易操作,在提高了烏苯美司產(chǎn)品的同時(shí),收率高;
(2)本發(fā)明的利用了dcc催化后先形成了活化酯溶液,做成活化酯后利用乙酸乙酯為溶劑,溶解性已經(jīng)可以滿足工藝要求,不需要采用四氫呋喃即可滿足溶解性,并且通過(guò)采用弱酸強(qiáng)堿無(wú)機(jī)鹽也一部分解決了溶解性的問(wèn)題,提高了溶解性,可見(jiàn)本發(fā)明采用的弱酸強(qiáng)堿無(wú)機(jī)鹽不僅起到了催化作用,也間接起到了溶解的作用,可見(jiàn)從工藝流程設(shè)計(jì)上也具有一定的創(chuàng)新性,先形成了活化酯降低了反應(yīng)體系的溶解性要求。
附圖說(shuō)明
為了更清楚地說(shuō)明本發(fā)明發(fā)明具體實(shí)施方式或現(xiàn)有技術(shù)中的技術(shù)方案,下面將對(duì)具體實(shí)施方式或現(xiàn)有技術(shù)描述中所需要使用的附圖作簡(jiǎn)單地介紹,顯而易見(jiàn)地,下面描述中的附圖是本發(fā)明發(fā)明的一些實(shí)施方式,對(duì)于本領(lǐng)域普通技術(shù)人員來(lái)講,在不付出創(chuàng)造性勞動(dòng)的前提下,還可以根據(jù)這些附圖獲得其他的附圖。
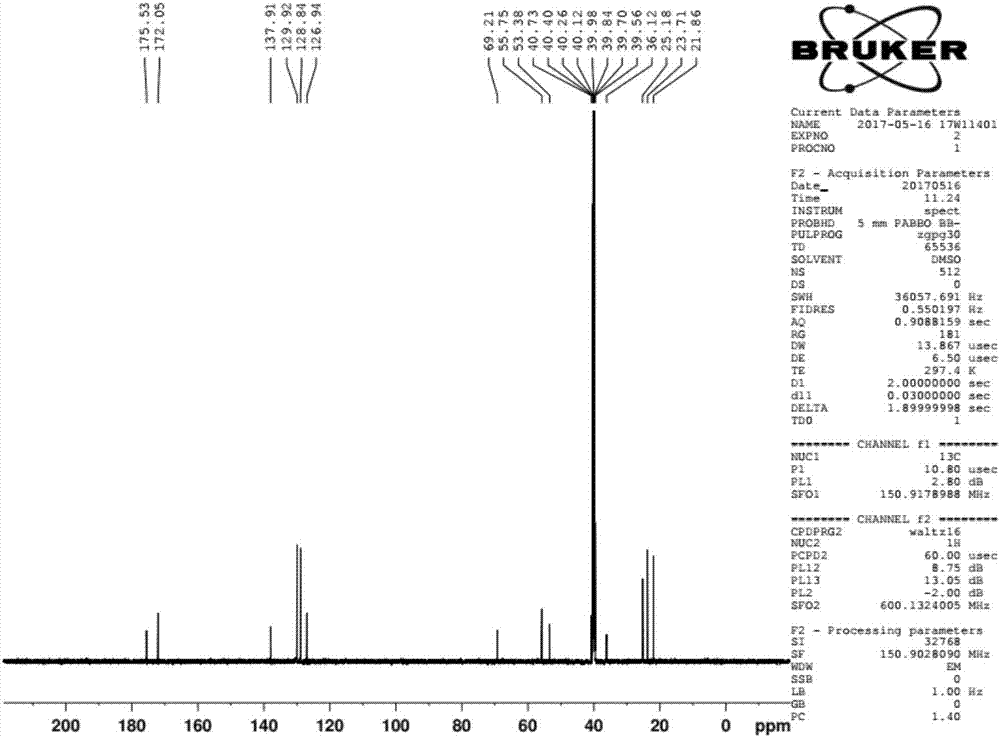
圖1-2為本發(fā)明實(shí)施例1中的烏苯美司產(chǎn)品的確認(rèn)圖譜。
具體實(shí)施方式
下面將結(jié)合實(shí)施例對(duì)本發(fā)明的實(shí)施方案進(jìn)行詳細(xì)描述,但是本領(lǐng)域技術(shù)人員將會(huì)理解,下列實(shí)施例僅用于說(shuō)明本發(fā)明,而不應(yīng)視為限制本發(fā)明的范圍。實(shí)施例中未注明具體條件者,按照常規(guī)條件或制造商建議的條件進(jìn)行。所用試劑或儀器未注明生產(chǎn)廠商者,均為可以通過(guò)市售購(gòu)買獲得的常規(guī)產(chǎn)品。
實(shí)施例1
烏苯美司的制備方法如下:
1)將l-亮氨酸芐酯對(duì)甲苯磺酸鹽、hobt、10倍(v/m)乙酸乙酯依次加入到反應(yīng)釜中,投料完成不斷攪拌降溫,緩慢加入dcc的乙酸乙酯溶液,加入過(guò)程體系溫度0-5℃之間。投料完成后體系在15-20℃間反應(yīng)4h,tlc跟蹤反應(yīng)終點(diǎn)(乙酸乙酯:石油醚=2:1、紫外254nm),得到l-亮氨酸芐酯對(duì)甲苯磺酸鹽的活化酯溶液;
2)將(2s,3r)-3-芐氧甲酰胺基-2-羥基-4-苯基丁酸、10倍(v/m)10%的碳酸氫鈉溶液依次加入到反應(yīng)釜中,攪拌至完全溶解,降溫至10-15℃,快速攪拌下滴加l-亮氨酸芐酯對(duì)甲苯磺酸鹽的活化酯溶液,滴加完成后在15-20℃間反應(yīng)4h左右,tlc跟蹤反應(yīng)終點(diǎn)(乙酸乙酯:石油醚=2:1、紫外254nm);
3)過(guò)濾分液,水相依次用7倍、2倍(v/m)乙酸乙酯萃取,合并乙酸乙酯相,乙酸乙酯相依次用6倍0.5wt%的鹽酸洗滌一次,6倍5wt%的飽和碳酸氫鈉洗滌一次,6倍純化水洗滌一次;乙酸乙酯溶液經(jīng)無(wú)水硫酸鈉干燥2h,過(guò)濾、濃縮至干,向濃縮物中加入5倍(v/m)石油醚,攪拌2h分散,過(guò)濾、40-45℃下干燥得n-[(2s,3r)-3-芐氧甲酰胺基-2-羥基-4-苯基丁酰]-l-亮氨酸,收率94-98%;
4)在室溫下將n-[(2s,3r)-3-芐氧甲酰胺基-2-羥基-4-苯基丁酰]-l-亮氨酸、pd/c(30%)溶解在15倍(v/m)的冰醋酸中,攪拌混合均勻后加入到反應(yīng)釜中,氮?dú)鈿錃庖来沃脫Q三次,1.5-2mpa下攪拌反應(yīng)至不吸氫為至;
5)過(guò)濾后的濾液在80℃、-0.1mpa下濃縮至干,濃縮物中加入10倍(v/m)丙酮,攪拌分散4h,過(guò)濾、40-45℃下鼓風(fēng)干燥得烏苯美司醋酸鹽;
6)將烏苯美司醋酸鹽溶解在鹽酸溶液中,溶液降溫至0-5℃之間,用15wt%的氨水調(diào)節(jié)溶液ph值至5-6。0-5℃下析晶5h,過(guò)濾,固體用丙酮打漿3h,過(guò)濾,40℃下干燥得到烏苯美司成品,收率為90.5%,具體確認(rèn)圖譜見(jiàn)附圖1-2。
實(shí)施例2
烏苯美司的制備方法如下:
1)將l-亮氨酸芐酯對(duì)甲苯磺酸鹽、hobt、10倍(v/m)乙酸乙酯依次加入到反應(yīng)釜中,投料完成不斷攪拌降溫,緩慢加入dcc的乙酸乙酯溶液,加入過(guò)程體系溫度0-5℃之間。投料完成后體系在15-20℃間反應(yīng)5h,tlc跟蹤反應(yīng)終點(diǎn)(乙酸乙酯:石油醚=2:1、紫外254nm),得到l-亮氨酸芐酯對(duì)甲苯磺酸鹽的活化酯溶液;
2)將(2s,3r)-3-芐氧甲酰胺基-2-羥基-4-苯基丁酸、10倍(v/m)20%的碳酸氫鈉溶液依次加入到反應(yīng)釜中,攪拌至完全溶解,降溫至10-15℃,快速攪拌下滴加l-亮氨酸芐酯對(duì)甲苯磺酸鹽的活化酯溶液,滴加完成后在15-20℃間反應(yīng)5h左右,tlc跟蹤反應(yīng)終點(diǎn)(乙酸乙酯:石油醚=2:1、紫外254nm);
3)過(guò)濾分液,水相依次用7倍、2倍(v/m)乙酸乙酯萃取,合并乙酸乙酯相,乙酸乙酯相依次用6倍0.5wt%的鹽酸洗滌一次,6倍5wt%的飽和碳酸氫鈉洗滌一次,6倍純化水洗滌一次;乙酸乙酯溶液經(jīng)無(wú)水硫酸鈉干燥2h,過(guò)濾、濃縮至干,向濃縮物中加入5倍(v/m)石油醚,攪拌3h分散,過(guò)濾、40-45℃下干燥得n-[(2s,3r)-3-芐氧甲酰胺基-2-羥基-4-苯基丁酰]-l-亮氨酸,收率94-98%;
4)在20-30℃條件下將n-[(2s,3r)-3-芐氧甲酰胺基-2-羥基-4-苯基丁酰]-l-亮氨酸、pd/c(30%)溶解在15倍(v/m)的冰醋酸中,攪拌混合均勻后加入到反應(yīng)釜中,氮?dú)鈿錃庖来沃脫Q三次,1.5-2mpa下攪拌反應(yīng)至不吸氫為至;
5)過(guò)濾后的濾液在70℃、-0.05mpa下濃縮至干,濃縮物中加入10倍(v/m)丙酮,攪拌分散5h,過(guò)濾、40-45℃下鼓風(fēng)干燥得烏苯美司醋酸鹽;
6)將烏苯美司醋酸鹽溶解在鹽酸溶液中,溶液降溫至0-5℃之間,用15wt%的氨水調(diào)節(jié)溶液ph值至5-6。0-5℃下析晶4h,過(guò)濾,固體用丙酮打漿3h,過(guò)濾,50℃下干燥得到烏苯美司成品,收率為92.3%。
實(shí)施例3
烏苯美司的制備方法如下:
1)將l-亮氨酸芐酯對(duì)甲苯磺酸鹽、hobt、10倍(v/m)乙酸乙酯依次加入到反應(yīng)釜中,投料完成不斷攪拌降溫,緩慢加入dcc的乙酸乙酯溶液,加入過(guò)程體系溫度0-5℃之間。投料完成后體系在15-20℃間反應(yīng)4h,tlc跟蹤反應(yīng)終點(diǎn)(乙酸乙酯:石油醚=2:1、紫外254nm),得到l-亮氨酸芐酯對(duì)甲苯磺酸鹽的活化酯溶液;
2)將(2s,3r)-3-芐氧甲酰胺基-2-羥基-4-苯基丁酸、10倍(v/m)20%的碳酸氫鈉溶液依次加入到反應(yīng)釜中,攪拌至完全溶解,降溫至10-15℃,快速攪拌下滴加l-亮氨酸芐酯對(duì)甲苯磺酸鹽的活化酯溶液,滴加完成后在15-20℃間反應(yīng)4h左右,tlc跟蹤反應(yīng)終點(diǎn)(乙酸乙酯:石油醚=2:1、紫外254nm);
3)過(guò)濾分液,水相依次用7倍、2倍(v/m)乙酸乙酯萃取,合并乙酸乙酯相,乙酸乙酯相依次用6倍0.5wt%的鹽酸洗滌一次,6倍5wt%的飽和碳酸氫鈉洗滌一次,6倍純化水洗滌一次;乙酸乙酯溶液經(jīng)無(wú)水硫酸鈉干燥2h,過(guò)濾、濃縮至干,向濃縮物中加入5倍(v/m)石油醚,攪拌2h分散,過(guò)濾、40-45℃下干燥得n-[(2s,3r)-3-芐氧甲酰胺基-2-羥基-4-苯基丁酰]-l-亮氨酸,收率94-98%;
4)在20-30℃條件下將n-[(2s,3r)-3-芐氧甲酰胺基-2-羥基-4-苯基丁酰]-l-亮氨酸、pd/c(30%)溶解在15倍(v/m)的冰醋酸中,攪拌混合均勻后加入到反應(yīng)釜中,氮?dú)鈿錃庖来沃脫Q三次,1.5-2mpa下攪拌反應(yīng)至不吸氫為至;
5)過(guò)濾后的濾液在75℃、-0.085mpa下濃縮至干,濃縮物中加入10倍(v/m)丙酮,攪拌分散4h,過(guò)濾、40-45℃下鼓風(fēng)干燥得烏苯美司醋酸鹽;
6)將烏苯美司醋酸鹽溶解在鹽酸溶液中,溶液降溫至0-5℃之間,用20wt%的氨水調(diào)節(jié)溶液ph值至5-6。0-5℃下析晶5h,過(guò)濾,固體用丙酮打漿2h,過(guò)濾,45℃下干燥得到烏苯美司成品,收率為91.4%。
比較例1
將(2s,3r)-3-芐氧甲酰胺基-2-羥基-4-苯基丁酸、l-亮氨酸芐酯對(duì)甲苯磺酸鹽、hobt、四氫呋喃溶依次加入到反應(yīng)釜中,投料完成體系攪拌下降溫到0℃左右,依次緩慢加入三乙胺和dcc,加入過(guò)程體系溫度不超過(guò)10℃。投料完成后體系在15℃~20℃間反應(yīng),攪拌反應(yīng)過(guò)夜,tlc跟蹤反應(yīng)終點(diǎn)(乙酸乙酯:石油醚=2:1、紫外254nm)。
過(guò)濾,濾液在45℃、-0.085mpa下減壓濃縮至干,得黃色油狀物濃縮物,加入15倍(v/m)乙酸乙酯溶解,溶液依次用4倍0.5n的鹽酸洗滌一次、4倍0.5n的飽和碳酸氫鈉洗滌一次、4倍純化水洗滌一次,乙酸乙酯溶液經(jīng)無(wú)水硫酸鈉干燥2小時(shí)。
過(guò)濾、濃縮至干,向濃縮物中加入10倍(v/m)石油醚,攪拌2小時(shí)分散,過(guò)濾、40℃~45℃下干燥得n-[(2s,3r)-3-芐氧甲酰胺基-2-羥基-4-苯基丁酰]-l-亮氨酸,收率80%-82%。
盡管已用具體實(shí)施例來(lái)說(shuō)明和描述了本發(fā)明,然而應(yīng)意識(shí)到,在不背離本發(fā)明的精神和范圍的情況下可以作出許多其它的更改和修改。因此,這意味著在所附權(quán)利要求中包括屬于本發(fā)明范圍內(nèi)的所有這些變化和修改。
- 還沒(méi)有人留言評(píng)論。精彩留言會(huì)獲得點(diǎn)贊!