R?琥珀酸曲格列汀光學純度的測定方法與流程
本發明涉及一種二肽基肽酶-Ⅳ(DPP-Ⅳ)抑制劑即R-琥珀酸曲格列汀光學純度的檢測方法。
背景技術:
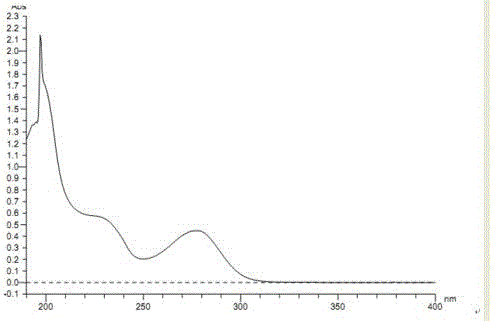
:琥珀酸曲格列汀(TrelagliptinSuccinate)是一種二肽基肽酶-Ⅳ(DPP-Ⅳ)抑制劑,其化學名為2-〔〔6-〔(3R)-3-氨基-1-哌啶基〕-3,4-二氫-3-甲基-2,4-二氧代-1(2H)-嘧啶基〕甲基〕-4-氟苯甲腈琥珀酸鹽。其主要成分為曲格列汀。該化合物結構中有一個手性中心,但真正具有藥理作用的為R構型,其結構式見式1:式1:琥珀酸曲格列汀結構式琥珀酸曲格列汀是由日本武田制藥開發的二肽基肽酶-Ⅳ(DPP-Ⅳ)抑制劑,通過選擇性、持續性抑制DPP-Ⅳ,從而控制血糖水平。腸促胰島素(GLP-Ⅰ)和葡萄糖依賴性促胰島素多肽(GIP)在血糖調節中發揮著重要的作用,而DPP-Ⅳ是能夠引發腸促胰島素(GLP-Ⅰ)和葡萄糖依賴性促胰島素多肽(GIP)的失活。通過抑制DPP-Ⅳ,就能夠增加血糖水平依賴性胰島素分泌,從而達到控制血糖的目的。琥珀酸曲格列汀是可通過提高機體自身能力控制血糖水平的新型的Ⅱ型糖尿病藥物,用作單藥,每周服用一次。其作用機制獨特,具有不產生低血糖、不引起體重增加,以及副作用小等獨特優勢,引起胃腸道不良反應的發生率也很低。2015年3月獲得日本厚生勞動省(MHLW)的上市批準,主要用于治療Ⅱ型糖尿病,耐受性良好。R-琥珀酸曲格列汀的合成是以3-甲基-6-氯尿嘧啶與2-氰基-5-氟溴芐反應生成2-〔(6-氯-3,4-二氫-3-甲基-2,4-二氧代-1(2H)-嘧啶基)甲基〕-4-氟苯甲腈,2-〔(6-氯-3,4-二氫-3-甲基-2,4-二氧代-1(2H)-嘧啶基)甲基〕-4-氟苯甲腈再與R-3-氨基哌啶雙鹽酸鹽反應生成R-曲格列汀,R-曲格列汀再與琥珀酸成鹽得到的R-琥珀酸曲格列汀。隨著手性液相分離技術的發展,高效液相色譜法已經成為能夠準確檢測物質光學純度的可靠的方法。但是R-琥珀酸曲格列汀的手性分離方法至今尚未見報道。究其原因,一是對映異構體雜質的色譜行為與主藥行為比較相似,必須選用特定的手性填料才能將二者徹底分離,需要進行大量的手性柱篩選實驗;二是由于其手性中心能夠與手性色譜柱填料產生氫鍵、偶極-偶極或空間作用的作用力較小,常規手性色譜柱的分離效能較低,無法實現有效的分離。為了有效的控制產品質量,迫切的需要一種快速、準確的檢測方法。本發明的目的:為了解決分離效能較低這一問題,我們開發了采用添加二乙胺的流動相的手性色譜法測定R-琥珀酸曲格列汀汀光學純度的方法。該方法是利用在流動相中添加二乙胺,調節流動相pH,穩定手性填料的空間構型,改變了手性填料對R-琥珀酸曲格列汀與S-琥珀酸曲格列汀的鍵合作用,從而提高了R-琥珀酸曲格列汀與S-琥珀酸曲格列汀之間的分離效能,實現了用手性色譜法進行光學純度的測定,首次創新性地解決了R-琥珀酸曲格列汀光學純度測定這一技術難題。技術實現要素:目前,琥珀酸曲格列汀按式2所示流程制備。式2琥珀酸曲格列汀的工藝流程本發明的技術方案:一種R-琥珀酸曲格列汀光學純度的測定方法,其特征在于,色譜條件:色譜系統為高效液相色譜儀,色譜柱填料為直鏈淀粉-三(3,5-二甲苯基氨基甲酸酯),即AD-H手性色譜柱(250mm×4.6mm,5μm);流動相A為正己烷,流動相B為二乙胺的乙醇溶液,且所述的正己烷與0.1%二乙胺的乙醇溶液的體積比為55:45~65:35,二乙胺的濃度為0.05%~0.15%,流動相在0.8~1.2mL·min-1,檢測波長:278nm。本發明技術方案中二乙胺的乙醇溶液中,二乙胺的作用是改性劑,通常加在正相的流動相中,主要的作用是調節流動相pH,穩定手性填料的空間構型,改變手性填料對R,S對映異構體的鍵合作用。所述流動相中二乙胺的乙醇溶液中,二乙胺的濃度為0.1%。所述流動相中正己烷與0.1%二乙胺的乙醇溶液的體積比是60:40。本發明的光學純度的檢測方法,其特征在于,含有以下步驟:取R-琥珀酸曲格列汀對照品、S-琥珀酸曲格列汀對照品各適量,分別置于25ml量瓶中,分別加無水乙醇溶解并定量稀釋制成各約為0.5mg·ml-1的溶液,作為R-琥珀酸曲格列汀對照品貯備液;(1)R,S-琥珀酸曲格列汀對照品溶液:精密量取上述貯備液各0.25ml,置同一100ml量瓶中,加無水乙醇稀釋至刻度,搖勻,即得,作為系統適應性試驗溶液;(2)S-琥珀酸曲格列汀對照品溶液:精密量取上述S-琥珀酸曲格列汀對照品貯備液0.25ml,置于100ml量瓶中,加無水乙醇稀釋至刻度,搖勻,即得,作為S-琥珀酸曲格列汀對照品定位溶液;(3)R-琥珀酸曲格列汀對照品溶液:精密量取上述R-琥珀酸曲格列汀對照品貯備液0.25ml,置于100ml量瓶中,加無水乙醇稀釋至刻度,搖勻,即得,作為R-琥珀酸曲格列汀對照品定位溶液;(4)R-琥珀酸曲格列汀供試品溶液:精密稱取R-琥珀酸曲格列汀供試品適量,加無水乙醇定量稀釋制成每1ml約含0.5mg的溶液,作為供試品溶液;(5)精密量取供試品溶液0.5ml,置200ml量瓶中,加無水乙醇稀釋至刻度,搖勻,作為對照溶液;(6)取對照溶液20μl注入液相色譜儀,調節檢測靈敏度,使主成分色譜峰的峰高約為滿量程的10%~20%。精密量取供試品溶液與對照溶液各20μl,分別注入液相色譜儀,記錄色譜圖至主成分峰保留時間的2倍,按峰面積歸一化法計算光學純度;色譜條件:以CHIRALPAKAD-H手性柱為色譜柱;以正己烷-無水乙醇(60:40)(含0.1%二乙胺)為流動相;檢測波長為278nm;柱溫為25℃;流速為每分鐘1.0mL,進樣量20μL。首先分別對R,S-琥珀酸曲格列汀對照品溶液與S-琥珀酸曲格列汀對照品溶液進樣,進行系統適應性分析,再對R-琥珀酸曲格列汀供試品溶液進行光學純度測定,按面積歸一化法計算樣品的光學純度。系統適應性試驗:R,S-琥珀酸曲格列汀達到了基線分離,分離度大于2.0,見說明書附圖3,與R-琥珀酸曲格列汀、S-琥珀酸曲格列汀對照品色譜圖(說明書附圖4與5)對比,確定RT=14.113為S構型,RT=15.773為R構型。本發明的有益成果:1在流動相中添加二乙胺,調節流動相pH,穩定手性填料的空間構型,改變手性填料對R,S-琥珀酸曲格列汀的鍵合作用,從而提高了分離效能。2被分析物中R,S-曲格列汀之間達到基線分離,可操作性強,分析方法專屬性、靈敏度與準確度高,能為保證藥品的安全、有效、質量可控提供強有力的依據。3主峰峰型良好,主峰前后雜質峰與主峰分離度良好,色譜條件的系統適應性良好。4該方法的平均回收率為101.52%,RSD為1.75%,完全滿足要求。本發明技術方案中,所述R-琥珀酸曲格列汀對照品與S-琥珀酸曲格列汀對照品的確認:色譜條件:色譜系統為高效液相色譜儀,色譜柱填料為十八烷基硅烷鍵合硅膠(250mm×4.6mm,5μm),流速:1.0ml·min-1,柱溫:35℃,檢測波長:278nm,進樣量20μL,流動相:以乙腈為流動相A,0.01mol·L-1的磷酸二氫鉀(用飽和氫氧化鉀溶液調節pH值至6.2)-乙腈(90:10)為流動相B,按下表程序進行梯度洗脫;-1-1時間(分鐘)流動相A(%)流動相B(%)019930208055851555.519960199純度測定:分別精密稱取R-琥珀酸曲格列汀對照品與S-琥珀酸曲格列汀對照品適量,加乙腈定量稀釋制成每1ml約含0.5mg的溶液,進樣檢測,按峰面積歸一化法計算純度,純度應大于99.0%;附圖說明:圖1:RS-琥珀酸曲格列汀純度測定色譜圖。圖2:RS-琥珀酸曲格列汀色譜峰光譜圖。圖3:RS-琥珀酸曲格列汀光學純度測定色譜圖。圖4:S-琥珀酸曲格列汀光學純度測定色譜圖。圖5:R-琥珀酸曲格列汀光學純度測定色譜圖。圖6:R-琥珀酸曲格列汀樣品(PTG141202批次供試品)光學純度測定色譜圖。圖注:EP/JP表示歐洲藥典和日本藥典高效液相色譜法分離度計算方法。具體實施方式:一下實施例用于說明本發明,但不作為對本發明的限制。實施例1R-琥珀酸曲格列汀樣品的光學純度的測定儀器與試劑:高效液相色譜儀(日本日立公司;賽默飛世爾科技有限公司);AD-H手性色譜柱(250mm×4.6mm,5μm;大賽璐藥物手性技術有限公司);恒溫水浴鍋(江蘇金壇科興儀器廠);BT-25S電子天平(瑞士,梅特勒公司)。DL-1000D超聲儀(上海之信儀器有限公司)。正己烷(色譜純,天津市彪士奇科技有限公司);無水乙醇(色譜純,天津市彪士奇科技有限公司);二乙胺(分析純,天津廣成化學試劑有限公司);R-琥珀酸曲格列汀對照品(純度≧99.9%,迪沙藥業集團藥物研究院);S-琥珀酸曲格列汀對照品(純度≧99.0%,迪沙藥業集團藥物研究院);R-琥珀酸曲格列汀樣品(PTG141202批次,純度≧99.0%,迪沙藥業集團藥物研究院)。我們通過大量實驗進行了流動相色譜條件的篩選:1.1流動相的選擇琥珀酸曲格列汀為極性較大的化合物,故選擇正己烷-無水乙醇體系為流動相。流動相的配比考察發現,適當增加正己烷的比例有利于提高分離度,但是正己烷比例較大會導致保留時間過長,不利于分析檢測。流動相中添加二乙胺,會使主峰峰型趨于尖峰,柱效增加,理論板數達到要求。通過對二乙胺添加量(0.05%、0.1%、0.15%)的系統優化顯示,1.0%的添加量使分離效能最佳,因而選擇0.1%的二乙胺。流速考察中發現,適當的降低流速也能夠提高分離效能,但是會導致分析時間過長,造成不必要的浪費。通過對流速(0.8mL·min-1、1.0mL·min-1、1.2mL·min-1)的系統優化顯示,1.0mL·min-1的流速條件使分離效能最佳,因而流速選擇1.0mL·min-1。1.2紫外波長的選擇:琥珀酸曲格列汀在流動相中的最大紫外吸收波長為278nm。由于琥珀酸曲格列汀與其對映異構體的理化性質基本一致,最大紫外吸收波長也是在278nm,因而選擇278nm作為對映異構體的檢測波長。1.3樣品濃度的選擇:比較濃度分別為0.3mg·mL-1、0.5mg·mL-1、1.0mg·mL-1的樣品的色譜圖,實驗結果顯示,0.3mg·mL-1的樣品峰高較低,含量較小的對映異構體未檢出;1.0mg·mL-1的樣品濃度太大,色譜柱有過載的跡象;0.5mg·mL-1濃度的樣品在峰高及峰面積方面均能滿足要求,因此確定0.5mg·mL-1為檢出樣品的濃度。1.4耐用性:通過對流動相比例、流速與柱溫的變化對方法進行了耐用性考察,結果發現,乙醇比例在45%-35%之間、流速在0.8-1.2mL·min-1之間發生微小變化時,對分離效能沒有影響,對映異構體均能檢出,且其含量沒有顯著變化。室溫在17~23℃之間變動時,對保留時間略有影響,對分離效能沒有影響。按技術方案所述檢測方法檢測R-琥珀酸曲格列汀的光學純度。樣品測定(PTG141202批次供試品)精密稱取PTG141202批次供試品適量,加無水乙醇定量稀釋制成每1ml約含0.5mg的溶液,作為供試品溶液;精密量取供試品溶液0.5ml,置200ml量瓶中,加無水乙醇稀釋至刻度,搖勻,作為對照溶液;取對照溶液20μl注入液相色譜儀,調節檢測靈敏度,使主成分色譜峰的峰高約為滿量程的10%~20%。精密量取供試品溶液與對照溶液各20μl,分別注入液相色譜儀,記錄色譜圖至主成分峰保留時間的2倍,按峰面積歸一化法計算光學純度(見附圖6);上述雖然結合附圖對本發明的具體實施方式進行了描述,但并非對本發明保護范圍的限制,所屬
技術領域:
技術人員應該明白,在本發明的技術方案的基礎上,本領域技術人員不需要付出創造性勞動即可做出的各種修改或變形仍在本發明的保護范圍以內。當前第1頁1 2 3
相關技術
網友詢問留言
已有0條留言
- 還沒有人留言評論。精彩留言會獲得點贊!
1