PM2.5切割器切割特性檢測用標準物質的定值方法及測量系統與流程
本發明涉及標準物質測量方法
技術領域:
,具體而言,涉及一種PM2.5切割器切割特性檢測用標準物質的定值方法及測量系統。
背景技術:
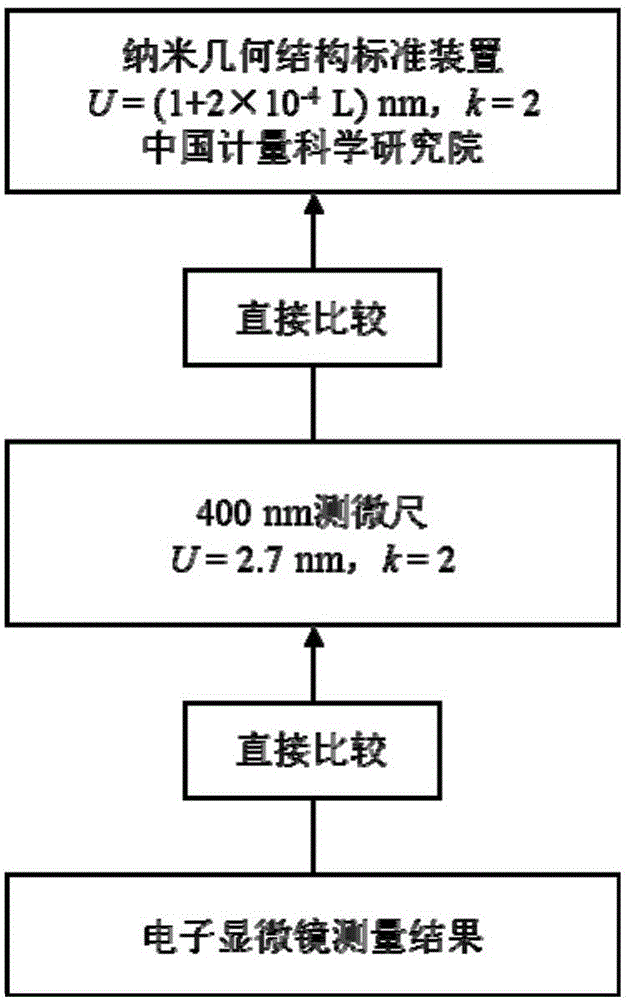
:PM2.5是指大氣中空氣動力學當量直徑小于或等于2.5μm的顆粒物,也稱為可入肺顆粒物。PM2.5粒徑小,富含大量的有毒、有害物質且在大氣中的停留時間長、輸送距離遠,因而對人體健康和大氣環境質量的影響更大。粒徑在2.5μm以下的細顆粒物,不易被阻擋,被吸入人體后會直接進入支氣管,干擾肺部的氣體交換,引發包括哮喘、支氣管炎和心血管病等方面的疾病。目前,PM2.5顆粒物是我國空氣首要的污染物,PM2.5監測及有效治理,是我國環境保護部門及國家政府的目標。2011年環境保護部頒布了HJ618-2011《環境空氣PM10和PM2.5的測定標準》,規定用于PM2.5顆粒物濃度監測的儀器為顆粒物采樣器和顆粒物直讀儀,按其原理可分為手工分析方法和自動分析方法(含:微量振蕩天平(TEOM)法、β射線測量法、光散射測量法)。無論是重量法、振蕩天平法、β射線法還是光散射測量法,PM2.5濃度測量均是通過切割器將大氣顆粒物中空氣動力學當量直徑小于或等于2.5μm的顆粒物分離出來,收集在濾膜上,然后進行測量分析。由此可以看出,PM2.5切割和濃度計量性能直接影響PM2.5監測儀的準確性,因此需要對PM2.5監測儀進行定期的檢校。對于PM2.5監測儀切割特性的檢測,美國國家環保局(EPA)標準《40CFRPart53,SubpartF》、我國國家環境保護標準HJ618-2011《環境空氣PM10和PM2.5的測定重量法》和日本JISZ8851:2008《大氣中PM2.5采樣器技術標準》中,都推薦使用標準物質(由不同空氣動力學當量直徑聚苯乙烯顆粒組成)對PM2.5監測儀切割特性進行檢測,檢測過程中需要的標準物質如表1所示。表1檢測PM2.5監測儀切割特性用標準物質要求表*表1中對標準物質要求為空氣動力學當量直徑,對單分散度無要求。由表1可以看出,PM2.5監測儀切割特性檢測需要使用8種空氣動力學當量粒徑范圍為(1.5-4.0)μm的標準物質。而目前,國內外的顆粒標準物質特性量值只有物理尺寸直徑,沒有針對PM2.5監測儀切割特性檢測用的空氣動力學標準物質,無法進行PM2.5監測儀切割特性檢測。導致這一現狀的原因在于空氣動力學當量直徑尚無法直接測量溯源,需通過顆粒的物理直徑與密度等量值進行轉換,其中存在諸多難點有待解決。有鑒于此,特提出本發明。技術實現要素:本發明的第一目的在于提供一種PM2.5切割器切割特性檢測用標準物質的定值方法,所述的PM2.5切割器切割特性檢測用標準物質的定值方法有效解決了定值過程中的空氣動力學直徑溯源、密度準確測量、不確定度評定以及適用性驗證等關鍵難點問題,是一種準確可靠的PM2.5切割器切割特性檢測用標準物質的測量方法。本發明的第二目的在于提供一種采用上述的PM2.5切割器切割特性檢測用標準物質的定值方法的測量系統,該系統能夠準確可靠地度對標準物質進行測定。為了實現本發明的上述目的,特采用以下技術方案:一種PM2.5切割器切割特性檢測用標準物質的定值方法,包括對所述標準物質顆粒空氣動力學當量直徑的定值、對所述標準物質顆粒空氣動力學當量直徑的定值不確定度的分析計算和對所述PM2.5切割器切割特性檢測用標準物質的定值方法的驗證。本發明PM2.5切割器切割特性檢測用標準物質的定值方法采用空氣動力學當量直徑對標準物質顆粒進行定值,并分析計算了對所述標準物質顆粒空氣動力學當量直徑的定值不確定度,還對其適用性進行了驗證,有效解決了定值過程中的空氣動力學直徑溯源、密度準確測量、不確定度評定以及適用性驗證等關鍵難點問題,是一種準確可靠的PM2.5切割器切割特性檢測用標準物質的定值方法。優選地,所述對標準物質顆粒空氣動力學當量直徑的定值通過對所述標準物質顆粒的物理直徑進行定值,然后按照如下公式計算得到:式中:Dp:顆粒的物理尺寸;Dae:顆粒空氣動力學當量直徑;CDP:顆粒物理直徑的坎寧安滑動修正因子,無量綱;CDae:顆粒空氣動力學當量直徑的坎寧安滑動修正因子,無量綱;ρ0:單位密度,ρ0=1.0g/cm3;ρp:氣溶膠顆粒密度,g/cm3。優選地,所述標準物質顆粒空氣動力學當量直徑的定值不確定度包括對所述標準物質的物理直徑進行定值引入的不確定度、對所述標準物質的氣溶膠密度進行測量引入的不確定度和所述標準物質的使用過程中引入的不確定度。優選地,對所述標準物質顆粒的物理直徑進行定值采用電遷移法、光學顯微鏡法、電子顯微鏡法或動態光散射法,優選采用電子顯微鏡法。進一步優選地,對所述標準物質顆粒的物理直徑進行定值選用與GBW(E)級標準物質相同的定值方法,即兩名操作人員使用絕對定值方法——掃描電子顯微鏡和圖像分析法對單分散粒度標準物質進行定值,利用量值可溯源至國家長度基準的400nm測微尺對掃描電子顯微鏡及圖像分析系統進行校準,之后再利用掃描電子顯微鏡對顆粒進行成像,圖像分析系統對顯微圖像進行處理和分析,最終確定顆粒的粒徑和粒徑分布;按JJF1006-1994《一級標準物質技術規范》的要求,兩名操作人員測量得到的數據經剔除異常值后檢驗平均值與標準偏差均無顯著性差異,之后將兩組數據合并,計算定值結果。優選地,所述氣溶膠顆粒密度包括所述標準物質顆粒真密度。優選地,對所述對標準物質顆粒真密度的測量采用與液體密度進行直接比較的方法,即將粉體材料分別放入一系列不同密度的液體中,觀察粉體材料在液體中的懸浮狀態以確定其密度;進一步優選地,對所述對標準物質顆粒真密度的測量采用JISZ89012006《實驗用粉體及試驗用粒子密度測量方法》。優選地,對所述標準物質的物理直徑進行定值引入的不確定度包括所述標準物質的物理直徑測量過程中引入的不確定度、所述標準物質均勻性引入不確定度和所述標準物質穩定性引入的不確定度。優選地,所述標準物質的物理直徑測量過程中引入的不確定度包括測微尺引入的不確定度、測量測微尺引入的不確定度和測量標準物質引入的不確定度。進一步優選地,所述測量測微尺引入的不確定度包括掃描電子顯微鏡分辨力引入的不確定度、測量重復性引入的不確定度、測微尺均勻性引入的不確定度、測量系統視野范圍內形變引入的不確定度和閾值設定引入的不確定度。進一步優選地,所述測量標準物質引入的不確定度包括掃描電子顯微鏡分辨力引入的不確定度、測量重復性引入的不確定度、視野范圍內的形變引入的不確定度和閾值設定引入的不確定度。優選地,對所述標準物質的氣溶膠密度進行測量引入的不確定度包括密度計測量準確性的不確定度、NaCl溶液判定的測量重復性不確定度和NaCl溶液密度變化間隔引入的不確定度。優選地,所述標準物質的使用過程中引入的不確定度包括使用溫度引入的不確定度和使用氣壓引入的不確定度。優選地,使用掃描電子顯微鏡和圖像分析的方法,對所述標準物質粒子進行均勻性檢驗。優選地,所述標準物質穩定性檢驗所采用的測量方法與其定值方法一致。優選地,對所述PM2.5切割器切割特性檢測用標準物質的定值方法的驗證包括采用標準物質對所述PM2.5切割器切割特性檢測用標準物質的測量方法準確性進行驗證、采用庫爾特粒度測量儀對所述標準物質顆粒空氣動力學當量直徑的定值進行測量和驗證、采用單顆粒氣溶膠質譜儀對所述標準物質顆粒空氣動力學當量直徑的定值進行測量和驗證,以及采用空氣動力學氣溶膠粒徑譜儀對所述標準物質顆粒空氣動力學當量直徑的定值進行測量和驗證中的一種或多種。采用上述的PM2.5切割器切割特性檢測用標準物質的測量方法的測量系統。本發明系統能夠準確可靠地度對標準物質進行定值。與現有技術相比,本發明的有益效果為:本發明PM2.5切割器切割特性檢測用標準物質的定值方法采用空氣動力學當量直徑對標準物質顆粒進行定值,并分析計算了對所述標準物質顆粒空氣動力學當量直徑的定值不確定度,還對其適用性進行了驗證,有效解決了定值過程中的空氣動力學直徑溯源、密度準確測量、不確定度評定以及適用性驗證等關鍵難點問題,是一種準確可靠的PM2.5切割器切割特性檢測用標準物質的定值方法。本發明系統能夠準確可靠地度對標準物質進行定值。附圖說明為了更清楚地說明本發明具體實施方式或現有技術中的技術方案,下面將對具體實施方式或現有技術描述中所需要使用的附圖作簡單地介紹,顯而易見地,下面描述中的附圖是本發明的一些實施方式,對于本領域普通技術人員來講,在不付出創造性勞動的前提下,還可以根據這些附圖獲得其他的附圖。圖1為本發明具體實施方式物理直徑量值溯源圖;圖2為本發明具體實施方式圖像分析處理流程圖;圖3為本發明具體實施方式密度量值溯源圖;圖4為本發明具體實施方式標稱值1.5μm市售聚苯乙烯標準物質的掃描電鏡照片;圖5為本發明具體實施方式標稱值1.5μm市售聚苯乙烯標準物質的粒徑分布圖;圖6為本發明具體實施方式標稱值2.0μm市售聚苯乙烯標準物質的掃描電鏡照片;圖7為本發明具體實施方式標稱值2.0μm市售聚苯乙烯標準物質的粒徑分布圖;圖8為本發明具體實施方式標稱值2.2μm市售聚苯乙烯標準物質的掃描電鏡照片;圖9為本發明具體實施方式標稱值2.2μm市售聚苯乙烯標準物質的粒徑分布圖;圖10為本發明具體實施方式標稱值2.5μm市售聚苯乙烯標準物質的掃描電鏡照片;圖11為本發明具體實施方式標稱值2.5μm市售聚苯乙烯標準物質的粒徑分布圖;圖12為本發明具體實施方式標稱值2.8μm市售聚苯乙烯標準物質的掃描電鏡照片;圖13為本發明具體實施方式標稱值2.8μm市售聚苯乙烯標準物質的粒徑分布圖;圖14為本發明具體實施方式標稱值3.0μm市售聚苯乙烯標準物質的掃描電鏡照片;圖15為本發明具體實施方式標稱值3.0μm市售聚苯乙烯標準物質的粒徑分布圖;圖16為本發明具體實施方式標稱值3.5μm市售聚苯乙烯標準物質的掃描電鏡照片;圖17為本發明具體實施方式標稱值3.5μm市售聚苯乙烯標準物質的粒徑分布圖;圖18為本發明具體實施方式標稱值4.0μm市售聚苯乙烯標準物質的掃描電鏡照片;圖19為本發明具體實施方式標稱值4.0μm市售聚苯乙烯標準物質的粒徑分布圖;圖20為本發明具體實施方式掃描電鏡視野內測量測微尺位置圖。具體實施方式下面將結合附圖和具體實施方式對本發明的技術方案進行清楚、完整地描述,但是本領域技術人員將會理解,下列所描述的實施例是本發明一部分實施例,而不是全部的實施例,僅用于說明本發明,而不應視為限制本發明的范圍。基于本發明中的實施例,本領域普通技術人員在沒有做出創造性勞動前提下所獲得的所有其他實施例,都屬于本發明保護的范圍。實施例中未注明具體條件者,按照常規條件或制造商建議的條件進行。所用試劑或儀器未注明生產廠商者,均為可以通過市售購買獲得的常規產品。在本發明的描述中,需要說明的是,術語“中心”、“上”、“下”、“左”、“右”、“豎直”、“水平”、“內”、“外”等指示的方位或位置關系為基于附圖所示的方位或位置關系,僅是為了便于描述本發明和簡化描述,而不是指示或暗示所指的裝置或元件必須具有特定的方位、以特定的方位構造和操作,因此不能理解為對本發明的限制。此外,術語“第一”、“第二”、“第三”僅用于描述目的,而不能理解為指示或暗示相對重要性。在本發明的描述中,需要說明的是,除非另有明確的規定和限定,術語“安裝”、“相連”、“連接”應做廣義理解,例如,可以是固定連接,也可以是可拆卸連接,或一體地連接;可以是機械連接,也可以是電連接;可以是直接相連,也可以通過中間媒介間接相連,可以是兩個元件內部的連通。對于本領域的普通技術人員而言,可以具體情況理解上述術語在本發明中的具體含義。本發明提供了一種PM2.5切割器切割特性檢測用標準物質的定值方法,包括對所述標準物質顆粒空氣動力學當量直徑的定值、對所述標準物質顆粒空氣動力學當量直徑的定值不確定度的分析計算和對所述PM2.5切割器切割特性檢測用標準物質的定值方法的驗證。優選地,所述對標準物質顆粒空氣動力學當量直徑的定值可通過對所述標準物質顆粒的物理直徑進行定值,然后按照如下公式計算得到:式中:Dp:顆粒的物理尺寸;Dae:顆粒空氣動力學當量直徑;CDP:顆粒物理直徑的坎寧安滑動修正因子,無量綱;CDae:顆粒空氣動力學當量直徑的坎寧安滑動修正因子,無量綱;ρ0:單位密度,ρ0=1.0g/cm3;ρp:氣溶膠顆粒密度,g/cm3。由上述公式可見,坎寧安滑動修正因子與物理直徑及空氣動力學當量直徑相關,無法推導空氣動力學當量直徑的解析式,因此需通過迭代計算的方法計算空氣動力學當量直徑。此外,坎寧安滑動修正因子亦受空氣溫度與氣壓的影響。坎寧安滑動修正因子的定義為:式中:l:空氣分子平均自由程;d:顆粒直徑;A1、A2、A3:由實驗確定的常數,具體數值為:A1=1.257,A2=0.400,A3=0.55。按硬球模型計算空氣分子的平均自由程:式中:μ:空氣粘度;p:氣壓;T:氣溫;M:空氣分子平均分子量,具體數值為:M=28.96g/mol。空氣粘度可按Sutherland公式計算:式中:λ:常數,λ=1.458μPa·s/K;C:Sutherland常數,C=110.4K。由公式(1-4)、(1-5)和(1-6)可見,坎寧安滑動修正因子隨溫度與氣壓的變化而變動,當環境溫度與壓力偏離室溫、1個大氣壓的條件時應考慮坎寧安滑動修正因子的變動對空氣動力學直徑轉換的影響。從以上計算公式可以看出:對于理想的球形顆粒,在已知其密度的情況下可由其物理直徑計算其在特定溫度與氣壓條件下的空氣動力學當量直徑,從而進行定值,但其中仍有三個關鍵難點問題需要解決:(1)密度的準確測量:由公式(1-1)可知,顆粒的球形度以及對其物理直徑與密度的準確測量直接關系到定值結果的準確度。微米級顆粒的球形度評價方法與物理直徑絕對定值方法已相對成熟,易于評價其球形度并對物理直徑進行準確定值,而其真密度的測量則不同于常規密度測量方法,對于少量顆粒的真密度進行準確測量尤為困難。(2)不確定度評定:由于物理直徑、密度、溫度與氣壓均可影響空氣動力學當量直徑的轉換,因此,對于空氣動力學當量直徑定值結果的不確定度評定需考慮上述各個因素的影響,加之空氣動力學當量直徑的轉換公式為非線性迭代計算模型,不確定度評定過程相當困難。(3)定值結果與適用性驗證:由于公式(1-1)-(1-3)僅適用于理想球形顆粒,且對于任意形狀的顆粒尚無統一的空氣動力學當量直徑計算公式,無法從理論上定量計算顆粒幾何形貌的隨機偏差對空氣動力學當量直徑的影響,因此雖然可通過球形度驗證確保顆粒的形貌滿足特定要求,但仍需使用基于空氣動力學測量原理的標準裝置驗證標準物質的定值結果及其適用性。本發明具體實施方式中所采用的標準物質顆粒為市售PM2.5鑒定用標準物質(GBW13642-GBW13649聚苯乙烯微球,生產商為北京普天同創生物科技有限公司),其空氣動力學當量直徑標稱值依次分別為1.5μm、2.0μm、2.2μm、2.5μm、2.8μm、3.0μm、3.5μm、4.0μm。下面采用本發明方法對市售PM2.5鑒定用標準物質進行空氣動力學當量直徑的定值、對所述標準物質顆粒空氣動力學當量直徑的定值不確定度的分析計算和對所述PM2.5切割器切割特性檢測用標準物質的測量方法的驗證。目前,國內外計量機構開展并研究了多種用于物理直徑的定值方法,主要有:電遷移法、光學顯微鏡法、電子顯微鏡法(透射和掃描電子顯微鏡)、動態光散射法等。氣溶膠電遷移法是NIST近年來新發展和研究的一種測量技術,但是到目前為止該測量方法的溯源性并沒有很好的解決。所以,利用該方法對標準物質進行定值時,需要利用現有的標準物質對該方法進行校準,該方法的測量結果溯源到已有的粒度標準物質。光子相關光譜法也是納米/亞微米級粒度標準物質的定值方法之一,該方法有復雜的數學計算和假設,需要精確測量環境溫度、液體黏度等參數,目前包括美國NIST、韓國KRISS在內的許多計量機構都在開展該方法測量原理及測量參數溯源的計量學研究。顯微鏡法(光學和電子顯微鏡)是顆粒粒徑測量方法中最權威和被廣泛認可的測量方法,測量結果可直接溯源到國家長度基準,是一種絕對定值方法。目前,國內外許多計量機構采用顯微鏡法對粒度標準物質進行定值時,常采用中心距離法、六方陣列法和單粒子測量法,如下所示:中心距離法的原理為:將含有待測顆粒的液體滴于顯微鏡的載波片上,烘干后由于表面張力的作用,顆粒間會形成線狀或串狀的兩兩接觸的結構。當用平行光照射時,每個透明的顆粒就象一個小的凸透鏡對入射的平行光具有折射的作用,并在其焦平面上聚焦成若干的小點,這些小點的位置正好代表了顆粒中心的位置。而相鄰兩個小點的距離C為兩個接觸顆粒的半徑之和。中心距離法的優點是可以不考慮顆粒的邊緣,從而減小了由于確定顆粒邊緣而造成的誤差和不確定度,但是該方法只適用于光學顯微鏡法。因此,鑒于光學顯微鏡的測量下限,該方法并不適于測量亞微米/納米級顆粒樣品,另外,該方法要求顆粒的均勻性性好,對于粒徑分布標準偏差較大的樣品該方法并不適合,存在較大的系統偏差。六方陣列法的原理為:將顆粒整齊排列后測量多個顆粒的總長度,從而確定單個顆粒的粒徑,NIST曾在八十年代采用六方陣列法對標稱值為1微米的粒度標準物質SRM1690進行了定值。使用該方法進行定值時,樣品顆粒的粒徑分布要求非常均一,否則當將顆粒排列為六方陣列時,顆粒間并不能真正地實現緊密排列而會產生間隙。這種間隙在50nm以下時很難被觀測到,從而被忽略。所以對于粒度分布標準偏差較大的樣品,該方法并不適用,存在較大的系統誤差。且研究表明六方整列法的測量不確定度比其他測量方法要大的多,因此NIST最終沒有采納六方陣列的定值數據。單粒子測量法就是采用不同的測試技術對樣品中的顆粒進行單獨測量,這是其與前兩種方法的顯著區別。該方法通過統計具有代表性顆粒數量的粒徑,最終確定樣品的平均粒徑。在此所采用的測試技術可分為直接測量和圖像分析。所謂直接測量就是利用顯微鏡中目鏡或物鏡標尺對顯微圖像中顆粒粒徑進行直接測量的技術。在該方法中由于顆粒邊緣效應,使得重復性不好,不同操作者測量的重現性差,且由于要統計較多的顆粒數量,從而使得該方法很費時。圖像分析法的測量原理為:被測量顆粒經顯微鏡顯微成像,并由CCD攝像機、圖像采集后,利用計算機對顆粒數字化圖像進行二值化處理,準確確定被分析顆粒的邊緣,之后系統再根據顆粒所占的面積計算得到顆粒的直徑。在顯微圖像分析法由于采用了先進的計算測量方法,使得該方法的測量重復性和重現性大大提高,同直接測量法相比具有高準確度、高精確度的特點。目前,國際標準化組織已在2004年公布了顆粒粒度分析-圖像分析方法的標準(ISO13322-1Particlesizeanalysis-ImageanalysismethodPart1:Staticimageanalysismethods)。在上述介紹的方法中,電子顯微鏡法(透射和掃描電子顯微鏡法)被認為是測量亞微米/微米級顆粒粒徑最權威和被廣泛認可的測量方法,測量結果可溯源至國家長度基準。因此,NIST在對其標準物質SRM1691、SRM1690、SRM1692定值時都選用了透射和掃描電子顯微鏡法作為定值方法,另外,韓國計量院KRISS也利用電子顯微鏡法對標準物質進行了定值研究。結果表明在5μm測量范圍內,電子顯微鏡的測量結果與其他測量方法(電遷移法、動態光散射法等)的準確度等級相當。其可以作為單分散粒度標準物質的定值方法。本發明選用與GBW(E)級標準物質相同的定值方法,即兩名操作人員使用絕對定值方法——掃描電子顯微鏡和圖像分析法對單分散粒度標準物質進行定值,利用量值可溯源至國家長度基準的400nm測微尺對掃描電子顯微鏡及圖像分析系統進行校準,之后再利用掃描電子顯微鏡對顆粒進行成像,圖像分析系統對顯微圖像進行處理和分析,最終確定顆粒的粒徑和粒徑分布;按JJF1006-1994《一級標準物質技術規范》的要求,兩名操作人員測量得到的數據經剔除異常值后檢驗平均值與標準偏差均無顯著性差異,之后將兩組數據合并,計算定值結果。物理直徑的量值溯源如圖1所示。將樣本均勻地覆蓋在Si片上,自然干燥。之后放于掃描電子顯微鏡的樣品室內,固定掃描電子顯微鏡的工作距離和加速電壓,用掃描電鏡拍攝樣品照片,每個樣本所拍攝的粒子個數不小于800個。被測物由掃描電子顯微鏡放大成像,并經CCD攝像機、圖像采集卡后形成數字化圖像,數字化圖像的基本單位是像素,而一個像素所表示的實際大小與電子顯微鏡的放大倍數、圖像分析系統分辨率密切相關,因此在測量過程中需要對測量系統進行校準。圖像分析系統測量顆粒粒徑的原理為:被測物由顯微鏡成像并輸出后,生成一幅數字化灰度圖像,數字化圖像中包括了對象物、背景。為了能將對象物從整個圖像中區別出來,首先要進行圖像分割,而最常用的方法是設定某一閾值θ,將灰度圖像的數據分成兩部分,即大于閾值θ的部分和小于閾值θ的部分,這種用灰度變換來研究灰度圖像的方法稱為圖像的二值化。二值化處理就是求閾值θ,從而把灰度圖像分成圖像物和背景兩個區域。二值化的閾值是把圖像和背景區分開的標尺,適當的閾值既要盡可能的保留圖像信息,又要消除背景和噪聲的干擾,這是選擇閾值的原則。閾值的選擇直接影響顆粒粒徑的測量準確性。當閾值確定后,就可以通過測量顆粒所占的像素數量計算顆粒的面積,并利用公式(6-2)計算得到顯微放大后的顆粒直徑,最后系統再根據公式(6-3)計算得到顆粒的直徑。A=Nδ2(6-1);式中:A:顯微圖像中顆粒所占面積;D:顯微圖像中顆粒直徑;δ2:顯微圖像中每一個像素的面積;N:顯微圖像中顆粒所占的像素數;B:待測顆粒的直徑;l:測微尺的長度;L:測微尺在一定放大倍數下的測量值。每個樣本標準粒子的數量平均直徑和粒徑分布的相對標準偏差按下列公式計算:公式(6-4)-(6-6)中,δ為標準偏差;Di為單個粒子的直徑;D為粒子的平均直徑;n為粒子數目;ε為粒徑分布的相對標準偏差(分散系數)。圖像分析處理過程如圖2所示。粉體真密度是粉體質量與其真體積之比,其真體積不包括存在于粉體顆粒內部的封閉空洞。目前,在粉體材料真密度的測量應用領域,根據測定介質的不同,粉體真密度的主要測定方法為浸液法與氣體容積法。浸液法是將一定質量的粉體材料浸入在易潤濕顆粒表面的浸液中,測定其所排除液體的體積,從而計算得到其密度。浸液法的主要操作形式有比重瓶法和懸吊法,其中,比重瓶法具有儀器簡單、操作方便等優點。該方法必須使用真空脫氣(加熱法或/和減壓法)以完全排除粉體材料顆粒之間夾雜的氣泡與表面吸附的氣體。同時,所使用的浸液應不能溶解粉體材料或與之發生化學反應。氣體容積法是以氣體取代液體測定試樣所排出的體積,如美國康塔儀器公司的Ultrafoam粉體真密度測量儀。此法排除了浸液法對試樣溶解或發生化學反應的可能性,具有不損壞試樣的優點,氣體容積法又分為定容積法與不定容積法。但這種方法需要的粉體量很大,測定時易受溫度的影響,還需注意漏氣問題。上述兩種粉體材料真密度的主要測量方法適用于大多數科研與生產場合,但這兩類方法均需要大量的粉體材料(g或mL級)以得到較為可靠的質量與體積量值,且不適用于粒徑在5μm以下的顆粒。而PM2.5切割器檢測所需的標準物質固體量和顆粒直徑均很小,難以采用上述兩種常用方法準確測量其密度。由于聚苯乙烯材料的密度與水接近,因此可通過與液體密度進行直接比較的方法測量所研制的標準物質顆粒的密度,即將粉體材料分別放入一系列不同密度的液體中,觀察粉體材料在液體中的懸浮狀態以確定其密度。相比上述兩類測量方法,該方法具有操作相對簡單,測量準確度較高的特點。采用這一原理的測量標準有日本工業標準JISZ89012006《實驗用粉體及試驗用粒子密度測量方法》。美國EPA和我國環境保護部均推薦采用的該標準測量聚苯乙烯等大氣顆粒物相關材料的真密度。由上述真密度測量方法介紹可知,液體密度比較法適用于本研究所研制的標準物質的密度測量。因此本研究采用美國EPA和我國環境保護部推薦采用的日本工業標準JISZ89012006《實驗用粉體及試驗用粒子密度測量方法》測量所研制的標準粒子的密度。其測量方法為:(1)在20℃條件下,配制一系列(一般是10個)與聚苯乙烯粒子密度相當的以0.002g/m3為間隔的NaCl溶液,分別加入100mL的量筒;使用標準密度儀準確測定每種NaCl溶液的密度。(2)取少量聚苯乙烯粒子的沉淀物分別加入(1)中10組不同密度的量筒中。用攪拌棒充分攪拌混合,放置在20℃的恒溫箱中,一個月之后觀察每一個量筒中聚苯乙烯顆粒的懸浮狀態。(3)粒子在NaCl溶液中有3種狀態:1.漂浮;2.沉淀;3.均勻懸浮。粒子在判斷:粒子在NaCl溶液中均勻懸浮時,表示測量的顆粒密度與溶液密度相等,此時NaCl溶液的密度即為聚苯乙烯粒子密度。密度定值方法的量值溯源如圖3所示。綜上所述,將根據定值的標準物質的物理直徑和測量出的標準物質的真密度,帶入公式(1-1)、(1-2)、(1-3)中,即可計算得到對應該標準物質在室溫、1個大氣壓的條件下的空氣動力學當量直徑。具體地,本發明具體實施方式采用了單一實驗室絕對定值方法:掃描電子顯微鏡和圖像分析法對標準物質的物理直徑進行定值。采用兩種不同原理的密度測量方法對標準物質的密度進行定值。上述市售PM2.5鑒定用標準物質的量,每種為20瓶(10mL/瓶,固含量1%-2%)。按照JJF1006-1994《一級標準物質技術規范》的規定,將樣品單元編號后,每種抽取15瓶作為測量樣品,將測量樣品超聲3min并充分搖勻后,采用分層隨機抽取法(考慮到樣品會發生沉降的原因),從每個測量樣品中抽取3個樣本。采用線距為400nm的光柵,測微尺的量值由中國計量科學研究院采用納米幾何結構標準裝置測量,可溯源至國家長度基準,測量值為400.3nm,不確定度為2.7nm(k=2)。采用日立3500N型掃描電子顯微鏡,該儀器的圖像線性失真度小、方法倍數的重復性好。采用美國標樂86-3000型圖像分析系統對光柵和標準物質的圖像進行分析研究。采用經檢定合格的奧地利產AntonPaarDMA35型密度測量儀(振蕩管法)與玻璃浮計式密度計對NaCl溶液的密度進行測量。按照上述定值方法并設定儀器程序,使用標稱值為400nm的測微尺對日立3500N型掃描電鏡的5000倍放大倍數進行校準,之后在相同的放大倍數下對標準粒子進行成像,最后利用圖像分析系統測量測微尺及標準粒子的大小,并計算物理粒徑大小和分布。按照上述方法測量出8種市售PM2.5鑒定用標準物質的真密度,最后用公式(1-1)、(1-2)、(1-3)計算其空氣動力學當量直徑。使用線距為400nm測微尺對掃描電子顯微鏡5000倍放大倍數進行校準。具體方法為:在水平和垂直方向將測微尺放在顯微鏡視野中央成像,使用圖像分析系統對數字化圖像進行測量,其中水平方向測量測微尺的20格,垂直方向測量測微尺的16格。然后選取測微尺的不同區域重復上述步驟30次,得到:水平方向像素數平均值:397.7。垂直方向像素數平均值:318.2(表2)。測量計算得到5000倍放大倍數下,數字化圖像中一個像素的實際大小為a:水平方向垂直方向那么數字化圖像中一個像素的實際大小a為20.13nm。表2放大倍數的校準數據按照上方法進行取樣和顯微鏡電子成像,圖4是部分成像照片,圖5是對800個顆粒粒徑測量后的粒徑分布圖。可以看出,所得到的標準粒子具有良好的球形度,且表面較光滑、無破損、無缺陷均勻,粒徑分布呈正態分布。如前所述,在圖像分析中閾值的選擇直接影響顆粒粒徑的測量準確性。在這里,采用ISO13322-1推薦使用的閾值半高度法(half-amplitudemethodofthresholdlevelsetting)確定顆粒的邊緣,即:在某顆粒邊緣區域內,在顆粒上、且盡量靠近邊緣的地方選定某一點,測量該點的灰度大小;再在背景處、且與上述選定點距離幾個像素處選定另外一點,測量該點的灰度大小,計算這兩個選定點的平均閾值作為柵格邊緣閾值。按照上述方法,最終確定顆粒的邊緣閾值。按照上述方法計算得到標稱值1.5μm標準物質的物理數量平均粒徑為1452.8nm即1.4528μm,見表3。按照上述方法計算得到顆粒粒徑分布的相對標準偏差為1.68%。表3標稱值1.5μm標準物質的數量平均粒徑定值結果按照上述方法測量標準物質的真密度,得出其測量值為1.050g/cm3。測量步驟為:(1)配置NaCl標準比重溶液。在20℃下,以超純水(電阻率為18.25MΩ·cm以上)為溶劑,分別配置10組不同密度梯度的NaCl溶液,密度范圍為1.040~1.058g/cm3。(2)在20℃下用密度儀測量NaCl溶液密度值。每組溶液重復測量10次。(3)取少許標準物質分別加入(1)中10組不同密度的量筒中,用攪拌棒充分攪拌混合,放置在20℃的恒溫箱中,一個月之后觀察聚苯乙烯顆粒的懸浮狀態,觀察到溶液6中的聚苯乙烯粒子是均勻懸浮的。觀察結果見表4。表4聚苯乙烯顆粒真密度測量(4)使用標準密度儀重復測量10次溶液6,以其測量結果的平均值作為本標準顆粒的真密度值。見表5。表5標準物質真密度測量結果表將物理直徑1452.8nm和標準物質真密度1.050g/cm3帶入公式(1-1)、(1-2)、(1-3)中,經迭代計算得到對應的標準物質的空氣動力學當量粒徑為1491nm。按照上述方法為標稱值為2.0μm標準物質定值。按照上述方法測量標準物質的真密度,得出其測量值為1.050g/cm3。圖6是部分成像照片,圖7是對800個顆粒粒徑測量后的粒徑分布圖。按照上述方法計算得到標稱值2.0μm標準物質的物理數量平均粒徑為1790.9nm,轉換為空氣動力學當量粒徑為1837nm,見表6。按照上述方法計算得到顆粒粒徑分布的相對標準偏差為1.79%。表6標稱值2.0μm標準物質的數量平均粒徑定值結果按照上述方法為標稱值2.2μm標準物質定值。按照上述測量標準物質的真密度,得出其測量值為1.050g/cm3。圖8是部分成像照片,圖9是對800個顆粒粒徑測量后的粒徑分布圖。按照上述方法計算得到標稱值2.2μm標準物質的物理數量平均粒徑為2161.3nm,轉換為空氣動力學當量粒徑為2217nm,見表7。按照上述方法計算得到顆粒粒徑分布的相對標準偏差為1.96%。表7標稱值2.2μm標準物質的數量平均粒徑定值結果按照上述方法為標稱值2.5μm標準物質定值。按照上述方法測量標準物質的真密度,得出其測量值為1.050g/cm3。圖10是部分成像照片,圖11是對800個顆粒粒徑測量后的粒徑分布圖。按照上述方法計算得到標稱值2.5μm標準物質的物理數量平均粒徑為2442.9nm,轉換為空氣動力學當量粒徑為2505nm,見表8。按照上述方法計算得到顆粒粒徑分布的相對標準偏差為2.89%。表8標稱值2.5μm標準物質的數量平均粒徑定值結果按照所述方法為標稱值2.8μm標準物質定值。按照所述方法測量標準物質的真密度,得出其測量值為1.050g/cm3。圖12是部分成像照片,圖13是對800個顆粒粒徑測量后的粒徑分布圖。按照所述方法計算得到標稱值2.8μm標準物質的物理數量平均粒徑為2743.6nm,轉換為空氣動力學當量粒徑為2813nm,見表9。按照所述方法計算得到顆粒粒徑分布的相對標準偏差為1.97%。表9標稱值2.8μm標準物質的數量平均粒徑定值結果按照上述方法為標稱值3.0μm標準物質定值。按照上述方法測量標準物質的真密度,得出其測量值為1.050g/cm3。圖14是部分成像照片,圖15是對800個顆粒粒徑測量后的粒徑分布圖。按照上述方法計算得到標稱值2.8μm標準物質的物理數量平均粒徑為3136.4nm,轉換為空氣動力學當量粒徑為3216nm,見表10。按照上述方法計算得到顆粒粒徑分布的相對標準偏差為1.81%。表10標稱值3.0μm標準物質的數量平均粒徑定值結果按照上述方法為3.0μm標準物質的定值。按照上述測量標準物質的真密度,得出其測量值為1.050g/cm3。圖16是部分成像照片,圖17是對800個顆粒粒徑測量后的粒徑分布圖。按照上述方法計算得到標稱值3.5μm標準物質的物理數量平均粒徑為3655.7nm,轉換為空氣動力學當量粒徑為3748nm,結果見表11。按照上述方法計算得到顆粒粒徑分布的相對標準偏差為1.35%。表11標稱值3.5μm標準物質的數量平均粒徑定值結果按照上述方法為4.0μm標準物質的定值。按照上述方法測量標準物質的真密度,得出其測量值為1.050g/cm3。圖18是部分成像照片,圖19是對800個顆粒粒徑測量后的粒徑分布圖。按照上述方法計算得到標稱值4.0μm標準物質的物理數量平均粒徑為4116.4nm,轉換為空氣動力學當量粒徑為4220nm,結果見表12。按照上述方法計算得到顆粒粒徑分布的相對標準偏差為1.58%。表12標稱值4.0μm標準物質的數量平均粒徑定值結果8種標準物質的空氣動力學當量直徑結果匯總如表13所示。表138種標準顆粒空氣動力學當量直徑結果匯總標稱直徑/μm1.52.02.22.52.83.03.54.0空氣動力學當量直徑/μm1.4911.8372.2172.5052.8133.2163.7484.220市售PM2.5鑒定用標準物質均勻性檢驗:使用掃描電子顯微鏡和圖像分析的方法,對市售PM2.5鑒定用標準物質粒子進行均勻性檢驗。該類標準物質用于PM2.5切割器切割特性校準試驗時,使用方法為將標準物質樣品滴入切割器特性檢測裝置的氣溶膠霧化發生器。因此該類標準物質在實際使用中的最小取樣量為1滴,約為0.05mL。在這里將樣品的最小取樣量定為0.05mL,進行均勻性、穩定性考察和定值研究。采用方差分析法(F檢驗法),此法是通過組間方差和組內方差的比較來判斷各組測量值之間有無系統偏差,即所謂方差分析的方法。如果二者的比小于統計檢驗的臨界值則認為樣品是均勻的。為檢驗樣品均勻性,設抽取了m個樣品,用重復性好的實驗方法,在相同條件下得m組等精度測量數據如下:平均值平均值…………………………………………;平均值設組間差方和組內差方和v1=m-1;v2=N-m;作統計量F:由此可以知道,該統計量是自由度為(v1,v2)的F分布變量。根據自由度(v1,v2)及給定的顯著性水平α,可由F分布臨界值表查得臨界的Fα值。若按公式(3-1)算得的F值有F<Fα,則認為組內與組間無明顯差異,樣品是均勻的。若F≥Fα,則懷疑各組間有系統誤差,即樣品之間存在差異。若記這個差異的均方根誤差為Sl,則若各ni均相同,均為n時,則公式(3-2)成為如下形式:均勻性檢驗結果:采用上述方法對標準物質的特性量值——數量平均粒徑進行均勻性統計,數據如下所示。表14標稱值為1.5μm標準物質的均勻性檢驗結果對于數量平均粒徑,計算得到組間差方和Q1=4.660,組內差方和Q2=9.000,其中υ1=14,υ2=30,那么計算得到:統計量F=1.109<F0.95(14,30)=2.04;則認為組內與組間無明顯差異,樣品是均勻的。表15標稱值為2.0μm標準物質的均勻性檢驗結果對于數量平均粒徑,計算得到組間差方和Q1=8.139,組內差方和Q2=10.20,其中υ1=14,υ2=30,那么計算得到:統計量F=1.710<F0.95(14,30)=2.04;則認為組內與組間無明顯差異,樣品是均勻的。表16標稱值為2.2μm標準物質的均勻性檢驗結果對于數量平均粒徑,計算得到組間差方和Q1=9.67,組內差方和Q2=17.85,其中υ1=14,υ2=30,那么計算得到:統計量F=1.16<F0.95(14,30)=2.04;則認為組內與組間無明顯差異,樣品是均勻的。表17標稱值為2.5μm標準物質的均勻性檢驗結果對于數量平均粒徑,計算得到組間差方和Q1=9.527,組內差方和Q2=11.55,其中υ1=14,υ2=30,那么計算得到:統計量F=1.767<F0.95(14,30)=2.04;則認為組內與組間無明顯差異,樣品是均勻的。表18標稱值為2.8μm標準物質的均勻性檢驗結果對于數量平均粒徑,計算得到組間差方和Q1=23.76,組內差方和Q2=28.58,其中υ1=14,υ2=30,那么計算得到:統計量F=1.782<F0.95(14,30)=2.04;則認為組內與組間無明顯差異,樣品是均勻的。表19標稱值為3.0μm標準物質的均勻性檢驗結果對于數量平均粒徑,計算得到組間差方和Q1=7.353,組內差方和Q2=13.93,其中υ1=14,υ2=30,那么計算得到:統計量F=1.131<F0.95(14,30)=2.04;則認為組內與組間無明顯差異,樣品是均勻的。表20標稱值為3.5μm標準物質的均勻性檢驗結果對于數量平均粒徑,計算得到組間差方和Q1=18.38,組內差方和Q2=32.00,其中υ1=14,υ2=30,那么計算得到:統計量F=1.23<F0.95(14,30)=2.04;則認為組內與組間無明顯差異,樣品是均勻的。表21標稱值為4.0μm標準物質的均勻性檢驗結果對于數量平均粒徑,計算得到組間差方和Q1=8.938,組內差方和Q2=14.30,其中υ1=14,υ2=30,那么計算得到:統計量F=1.339<F0.95(14,30)=2.04;則認為組內與組間無明顯差異,樣品是均勻的。市售PM2.5鑒定用標準物質均勻性檢驗:市售PM2.5鑒定用標準物質穩定性檢驗所采用的測量方法與其定值方法一致。長期穩定性試驗數據起止時間為2013.11-2014.11;短期穩定性試驗數據考察標準物質置于35℃環境中連續4周的粒徑變化。取樣方式及最小取樣量同3.1中的方法。采用ISOGuide35:2006中的趨勢檢驗法評價標準物質的穩定性,即對測量平均值與時間進行線性回歸,按方差分析表(表22)計算統計量F,并通過F-檢驗以判斷線性回歸的顯著性。表22線性回歸的方差分析表其中:Yi:任一次標準物質穩定性檢驗的測量平均值;全部標準物質穩定性檢驗的測量平均值;任一次標準物質穩定性檢驗的回歸曲線擬合值;n:測量次數。若統計量F<Fα(1,n-2),說明標準物質數量平均粒徑的測量值與時間的線性回歸是不顯著的,即該標準物質的測量值與認定值(標準值)一致,該標準物質的特性量值在考察時間段內沒有發生顯著性變化;否則認為該標準物質的特性量值發生了顯著性變化。分別采用上述方法對標稱值為1.5μm、2.0μm、2.2μm、2.5μm、2.8μm、3.0μm、3.5μm、4.0μm的市售PM2.5鑒定用標準物質的特性量值——數量平均粒徑進行測量,并評價標準物質的穩定性。長期穩定性檢驗結果:表23標稱值1.5μm標準物質的穩定性考察數據表24線性回歸的方差分析表可以看到統計量F<F0.95(1,2)=18.51,認為該標準物質的測量值與認定值(標準值)一致,該標準物質的特性量值沒有發生顯著性變化。表25標稱值2.0μm標準物質的穩定性考察數據表26線性回歸的方差分析表可以看到統計量F<F0.95(1,2)=18.51,認為該標準物質的測量值與認定值(標準值)一致,該標準物質的特性量值沒有發生顯著性變化。表27標稱值2.2μm標準物質的穩定性考察數據表28線性回歸的方差分析表可以看到統計量F<F0.95(1,2)=18.51,認為該標準物質的測量值與認定值(標準值)一致,該標準物質的特性量值沒有發生顯著性變化。表29標稱值2.5μm標準物質的穩定性考察數據表30線性回歸的方差分析表可以看到統計量F<F0.95(1,2)=18.51,認為該標準物質的測量值與認定值(標準值)一致,該標準物質的特性量值沒有發生顯著性變化。表31標稱值2.8μm標準物質的穩定性考察數據表32線性回歸的方差分析表可以看到統計量F<F0.95(1,2)=18.51,認為該標準物質的測量值與認定值(標準值)一致,該標準物質的特性量值沒有發生顯著性變化。表33標稱值3.0μm標準物質的穩定性考察數據表34線性回歸的方差分析表可以看到統計量F<F0.95(1,2)=18.51,認為該標準物質的測量值與認定值(標準值)一致,該標準物質的特性量值沒有發生顯著性變化。表35標稱值3.5μm標準物質的穩定性考察數據表36線性回歸的方差分析表可以看到統計量F<F0.95(1,2)=18.51,認為該標準物質的測量值與認定值(標準值)一致,該標準物質的特性量值沒有發生顯著性變化。表37標稱值4.0μm標準物質的穩定性考察數據表38線性回歸的方差分析表可以看到統計量F<F0.95(1,2)=18.51,認為該標準物質的測量值與認定值(標準值)一致,該標準物質的特性量值沒有發生顯著性變化。短期穩定性檢驗結果:表39標稱值1.5μm標準物質的穩定性考察數據表40線性回歸的方差分析表可以看到統計量F<F0.95(1,3)=10.13,認為該標準物質的測量值與認定值(標準值)一致,該標準物質的特性量值沒有發生顯著性變化。表41標稱值2.0μm標準物質的穩定性考察數據表42線性回歸的方差分析表可以看到統計量F<F0.95(1,3)=10.13,認為該標準物質的測量值與認定值(標準值)一致,該標準物質的特性量值沒有發生顯著性變化。表43標稱值2.2μm標準物質的穩定性考察數據表44線性回歸的方差分析表可以看到統計量F<F0.95(1,3)=10.13,認為該標準物質的測量值與認定值(標準值)一致,該標準物質的特性量值沒有發生顯著性變化。表45標稱值2.5μm標準物質的穩定性考察數據表46線性回歸的方差分析表可以看到統計量F<F0.95(1,3)=10.13,認為該標準物質的測量值與認定值(標準值)一致,該標準物質的特性量值沒有發生顯著性變化。表47標稱值2.8μm標準物質的穩定性考察數據表48線性回歸的方差分析表可以看到統計量F<F0.95(1,3)=10.13,認為該標準物質的測量值與認定值(標準值)一致,該標準物質的特性量值沒有發生顯著性變化。表49標稱值3.0μm標準物質的穩定性考察數據表50線性回歸的方差分析表可以看到統計量F<F0.95(1,3)=10.13,認為該標準物質的測量值與認定值(標準值)一致,該標準物質的特性量值沒有發生顯著性變化。表51標稱值3.5μm標準物質的穩定性考察數據表52線性回歸的方差分析表可以看到統計量F<F0.95(1,3)=10.13,認為該標準物質的測量值與認定值(標準值)一致,該標準物質的特性量值沒有發生顯著性變化。表53標稱值4.0μm標準物質的穩定性考察數據表54線性回歸的方差分析表可以看到統計量F<F0.95(1,3)=10.13,認為該標準物質的測量值與認定值(標準值)一致,該標準物質的特性量值沒有發生顯著性變化。所述市售PM2.5鑒定用標準物質可作為理想球體來處理,則其空氣動力學當量直徑可通過公式(1-1)-(1-3)計算。其中,公式(1-2)與(1-3)所給出的坎寧安滑動修正因子公式中的數值受空氣分子自由程影響,隨溫度與氣壓的變化而變動。由公式(1-1)可以看出,空氣動力學當量直徑的不確定度來源包括物理直徑引入的不確定度、密度引入的不確定度和使用過程中引入的不確定度(溫度和壓力的變化引入的不確定度)。根據顆粒物理直徑測量數學模型(公式8-1)分析,顆粒物理直徑引入的不確定度來源如表55所示。其中,D:待測顆粒的直徑;A:測微尺的標準長度;L:測微尺在一定放大倍數下的測量值;B:待測顆粒在一定放大倍數下的測量值。表55定值過程的不確定度來源400nm測微尺定值引入的不確定度:定值過程中使用的400nm測微尺的量值由中國計量科學研究院采用633nm激光穩頻裝置測量,可溯源至國家長度基準,測量值為A=400.3nm,測量不確定度均為2.7nm(k=2)。因此,放大5000倍數下測量400nm測微尺長度引入的不確定度:電鏡分辨力引入的不確定度:所謂分辨力,就是可以分辨的最小的兩點之間的間距。由于測量所使用的掃描電鏡直接輸出數字化的圖像,因而掃描電鏡的分辨力不確定度主要有兩部分來源:電鏡系統與成像系統。電鏡系統的分辨力取決于掃描電鏡的性能和調試達到的狀態,分辨率與電子束斑直徑、入射電子在試樣中的擴散、信噪比等因素相關,其中束斑直徑是主要影響因素。而在成像系統中,圖像傳感器上的每一個成像元件接收其所對應的尺度范圍內的信號,并輸入一個平均的信號強度,從而進一步引入了不確定度。在無法確定電鏡內部各組件技術參數的情況下,無法通過分析計算評價電鏡分辨力引入的不確定度,必須通過樣品測量進行評價。而樣品測量過程亦會引入額外的不確定度,即樣品測量結果的不確定度為電鏡分辨力與測量重復性、樣品均勻性、視野形變等不確定度的合成。故在此不單獨評價電鏡分辨力的不確定度,而認為其已并入其它各個測量過程評價得到的不確定度。測量測微尺重復性引入的不確定度δ21:在掃描電子顯微鏡成像及圖像分析過程中,儀器自身穩定性及一些操作都會影響到測量的重復性,如儀器加速電壓的漂移、樣品裝配不當導致工作面不垂直于電子光軸,工作距離漂移等,會對于實際的定值測量產生影響并引入不確定度。因此,我們考察了測量重復性對定值結果的影響。具體方法為:使用掃描電子顯微鏡在其5000倍放大倍數下,在水平和垂直方向對測微尺進行成像(視野范圍中央),并使用圖像分析系統對數字化圖像進行測量,其中水平方向測量測微尺的前20格,垂直方向測量測微尺的前16格記錄其結果。之后將測微尺從樣品室內取出,重復上述步驟10次,數據見表55。表56測微尺測量重復性的測量數據計算得到水平方向測量重復性為0.131%,垂直方向測量重復性為0.132%,因此得到測量重復性引入的不確定度δ21=0.132%。400nm測微尺均勻性引入的不確定度δ22:400nm測微尺的量值是所有柵格的平均值,而我們在對顯微鏡5000放大倍數進行校準時所用到的只是測微尺的某一些柵格,柵格間的均勻性會影響最終的測量結果。因此,我們考察了柵格的均勻性。具體方法為:在水平方向將測微尺放在顯微鏡視野中央成像,使用圖像分析系統對數字化圖像進行測量,分別測量測微尺的20個柵格和16個柵格的長度。然后選取測微尺的不同區域重復上述步驟30次,測量數據見表56。表57測微尺柵格均勻性的測量數據因此可以計算得到均勻性引入的不確定度δ22:測量系統視野范圍的形變引入的不確定度δ23:掃描電子顯微鏡的放大倍數、成像系統在其整個視野范圍內并不是一致的,而會存在一定的形變。因此,為了考察視野范圍內形變對測量結果引入的不確定度。將400nm測微尺的某2柵格在整個視野范圍的不同位置如圖20所示(左上、中上、右上、左中、中中、右中、左下、中下、右下)成像并測量,測量結果如表58所示,計算得到掃描電鏡的視野變形為0.56%,即:δ23=0.56%;表58放大5000倍數下視野范圍內的形變閾值設定引入的不確定度δ24:數字化的灰度圖像包括對象物和背景,從數字灰度圖像中提取出對象物,最常用的方法是設定某一閾值θ,將灰度圖像的數據分成兩部分,即大于閾值θ的部分和小于閾值θ的部分,這種用灰度變換來研究灰度圖像的方法稱為圖像的二值化。二值化處理就是求閾值θ,從而把灰度圖像分成圖像物和背景兩個區域。在400nm測微尺數字化圖像的灰度強度圖中測微尺邊緣與背景間灰度變化很大且很快,在邊緣區域內,從明顯的背景灰度點和柵格灰度點只經過1個像素點的過渡。因此一定范圍內不同的閾值設定最多可導致測微尺的邊緣產生1個像素的差別。但使用測微尺進行放大倍率校準時,測量的量值為測微尺柵格同側邊緣的距離,即閾值設定可導致距離測量結果產生±1個像素的變化。假設該變化為平均分布,則以16個柵格長度校準電子顯微鏡放大倍率時所引入的不確定度為:因此,通過以上的分析計算得到測量測微尺引入的不確定度為:在放大5000倍數下測量顆粒引入的不確定度;電鏡分辨力引入的不確定度;電鏡分辨力引入的不確定度不單獨評價,視為已合并入其它測量環節的不確定度評價過程。測量重復性引入的不確定度δ31:在掃描電子顯微鏡成像及圖像分析過程中,儀器自身穩定性及一些操作都會影響到測量的重復性,因此,我們考察了測量重復性對定值結果的影響,具體方法為:使用掃描電子顯微鏡在其5000倍放大倍數下,將某一顆粒進行成像(視野范圍中央),并使用圖像分析系統對數字化圖像進行測量。重復上述步驟10次,數據見表58。表59顆粒測量重復性的測量數據經計算得到測量重復性引入不確定度為:δ31=0.45%;測量系統視野范圍的形變引入的不確定度δ32:將同一顆粒在視野范圍內的不同位置處進行成像并測量,數據如表60所示,計算得到掃描電鏡視野形變的不確定度為0.32個像素,即δ32=0.44%。表605000放大倍數下視野范圍內的形變閾值設定引入的不確定度δ33:采用ISO13322-1推薦的閾值半高度法(half-amplitudemethodofthresholdlevelsetting)確定閾值。即:在明顯的顆粒邊緣處測量該點的閾值大小,再在遠離幾個像素處測量該點的閾值大小,計算平均閾值作為顆粒邊緣的閾值。為了考察顆粒邊緣灰度閾值變化對顆粒粒徑測量準確性的影響,將閾值在一定范圍內改變后看到,當閾值在110-121之間變換時,顯微圖像中背景干凈且顆粒邊緣規則,說明在閾值這個范圍內,顆粒粒徑測量準確。計算得到在這一閾值設定范圍內粒徑測量的標準偏差為0.4個像素,即閾值設定引入的不確定度δ33=0.55%。表61不同閾值設定所測量的粒徑值因此,通過以上的分析計算得到在5000放大倍數下測量標準物質引入不確定度為:測量顆粒引入不確定度的合成:由公式(8-1)得到:ur2(RM)=Ur2(A)+Ur2(B)+Ur2(L)(8-2);以空氣動力學標稱直徑為1.5μm顆粒為例,將上述分析的各項不確定分量代入公式(8-2)中得到顆粒測量過程中引入的不確定度:u(RM)=16.7nm;均勻性引入的不確定度評定根據《一級標準物質技術規范》(JJF1006-94)、《標準物質定值的通用原則及統計學原理》(JJF1343-2012)和《標準物質/標準樣品定值的一般原則和統計方法》(GB/T15000.3-2008)中的要求,對標準顆粒的均勻性引起的不確定度進行評定:標稱值1.5μm均勻性引起的不確定度:由均勻性檢驗結果知:由于因此按下式估算由顆粒的不均勻性引入的不確定度:SH=0.105nm;標稱值2.0μm均勻性引起的不確定度:由均勻性檢驗結果知:由于因此按下式估算由顆粒的不均勻性引入的不確定度:SH=0.284nm;標稱值2.2μm均勻性引起的不確定度:由均勻性檢驗結果知:由于因此按下式估算由顆粒的不均勻性引入的不確定度:SH=0.178nm;標稱值2.5μm均勻性引起的不確定度:由均勻性檢驗結果知:由于因此按下式估算由顆粒的不均勻性引入的不確定度:SH=0.314nm;標稱值2.8μm均勻性引起的不確定度由均勻性檢驗結果知:由于因此按下式估算由顆粒的不均勻性引入的不確定度:SH=0.486nm;標稱值3.0μm均勻性引起的不確定度:由均勻性檢驗結果知:由于因此按下式估算由顆粒的不均勻性引入的不確定度:SH=0.143nm;標稱值3.5μm均勻性引起的不確定度:由均勻性檢驗結果知:由于因此按下式估算由顆粒的不均勻性引入的不確定度:SH=0.191nm;標稱值4.0μm均勻性引起的不確定度:由均勻性檢驗結果知:由于因此按下式估算由顆粒的不均勻性引入的不確定度:SH=0.232nm;穩定性引入的不確定度評定:按ISOGuide35:2006采用趨勢分析的方法,對穩定性數據進行檢驗。標稱值1.5μm穩定性引起的不確定度:由穩定性檢驗結果知,以x代表穩定時間,以y代表顆粒直徑,擬合成一條直線。組數n=4。平均值為:月;直線的斜率:nm/月;直線的截距為:直線的標準偏差由下式計算:得直線的標準偏差:s=0.061nm;斜率的不確定度為:nm/月;則有效期t=12個月的長期穩定性的不確定度貢獻為:ST=Sb1·t=0.082nm。標稱值2.0μm穩定性引起的不確定度:由穩定性檢驗結果知,以x代表穩定時間,以y代表顆粒直徑,擬合成一條直線。組數n=4。平均值為:月;直線的斜率:nm/月;直線的截距為:直線的標準偏差由下式計算:得直線的標準偏差:s=0.059nm;斜率的不確定度為:nm/月;則有效期t=12個月的長期穩定性的不確定度貢獻為:ST=Sb1·t=0.079nm。標稱值2.2μm穩定性引起的不確定度:由穩定性檢驗結果知,以x代表穩定時間,以y代表顆粒直徑,擬合成一條直線。組數n=4。平均值為:月;直線的斜率:nm/月;直線的截距為:直線的標準偏差由下式計算:得直線的標準偏差:s=0.097nm;斜率的不確定度為:nm/月;則有效期t=12個月的長期穩定性的不確定度貢獻為:ST=Sb1·t=0.13nm。標稱值2.5μm穩定性引起的不確定度:由穩定性檢驗結果知,以x代表穩定時間,以y代表顆粒直徑,擬合成一條直線。組數n=4。平均值為:月;直線的斜率:nm/月;直線的截距為:直線的標準偏差由下式計算:得直線的標準偏差:s=0.047nm;斜率的不確定度為:nm/月;則有效期t=12個月的長期穩定性的不確定度貢獻為:ST=Sb1·t=0.063nm。標稱值2.8μm穩定性引起的不確定度:由穩定性檢驗結果知,以x代表穩定時間,以y代表顆粒直徑,擬合成一條直線。組數n=4。平均值為:月;直線的斜率:nm/月;直線的截距為:直線的標準偏差由下式計算:得直線的標準偏差:s=0.53nm;斜率的不確定度為:nm/月;則有效期t=12個月的長期穩定性的不確定度貢獻為:ST=Sb1·t=0.72nm。標稱值3.0μm穩定性引起的不確定度:由穩定性檢驗結果知,以x代表穩定時間,以y代表顆粒直徑,擬合成一條直線。組數n=4。平均值為:月;直線的斜率:nm/月;直線的截距為:直線的標準偏差由下式計算:得直線的標準偏差:s=0.084nm;斜率的不確定度為:nm/月;則有效期t=12個月的長期穩定性的不確定度貢獻為:ST=Sb1·t=0.12nm。標稱值3.5μm穩定性引起的不確定度:由穩定性檢驗結果知,以x代表穩定時間,以y代表顆粒直徑,擬合成一條直線。組數n=4。平均值為:月;直線的斜率:nm/月;直線的截距為:直線的標準偏差由下式計算:得直線的標準偏差:s=0.059nm;斜率的不確定度為:nm/月;則有效期t=12個月的長期穩定性的不確定度貢獻為:ST=Sb1·t=0.079nm。標稱值4.0μm穩定性引起的不確定度:由穩定性檢驗結果知,以x代表穩定時間,以y代表顆粒直徑,擬合成一條直線。組數n=4。平均值為:月;直線的斜率:nm/月;直線的截距為:直線的標準偏差由下式計算:得直線的標準偏差:s=0.076nm;斜率的不確定度為:nm/月;則有效期t=12個月的長期穩定性的不確定度貢獻為:ST=Sb1·t=0.10nm。物理直徑不確定度的合成:根據JJF1006-1994《一級標準物質技術規范》,由于各分量之間不相關,以標稱值為1.5μm顆粒為例,物理直徑合成標準不確定度為:uc(Dp)=16.9nm;ur(Dp)=1.17%;氣溶膠密度測量的不確定度分析:我們采用日本工業標準JISZ89012006《實驗用粉體及試驗用粒子密度測量方法》,測量所研制的標準顆粒的真密度。密度計測量準確性的不確定度:由HXYH13-1853計量檢測報告得知:DMA35液體密度計示值誤差為0.0006g/cm3.則密度引入的標準不確定度:NaCl溶液判定的測量重復性不確定度:根據結果選定第6組的密度為顆粒的真密度,重復測量的結果如表62所示。表62第6組重復測量結果表62為NaCl溶液10次(n=10)重復測量結果,按A類評定:單次測量結果的標準偏差:標準不確定度為:相對標準不確定度為:NaCl溶液密度變化間隔引入的不確定度:由于標準物質的真密度是與NaCl溶液密度比較得到,各組NaCl溶液的密度并非連續變化,其差值為0.002g/cm3,因此導致密度測量存在0.002g/cm3的分辨率,由此引入的不確定度為:綜上,氣溶膠密度測量總的不確定度為:使用過程引入的不確定度計算:空氣動力學的轉換公式(1-2)與(1-3)為坎寧安滑動修正因子在室溫、1個大氣壓下的計算式。根據公式(1-4)-(1-6)可知,該修正因子受氣溫與氣壓影響,因而溫度與氣壓可影響物理直徑至空氣動力學當量直徑的轉換結果。由于進行PM2.5監測儀切割特性測量時未控制氣溫與氣壓條件,故在不同地點、不同時間進行測量時,氣溫與氣壓的改變也引入了相應的不確定度。氣溫與氣壓所引入的不確定度體現為使用環節引入的不確定度,作為B類不確定度處理。考慮到相關計算過程復雜,難以推導不確定度的解析式,故氣溫與氣壓引入的不確定度通過數值計算得到。由于進行PM2.5切割器切割特性測量時,實驗室內的溫度波動范圍可控,而氣壓難以控制,因此將溫度范圍選定為(10-40)℃,氣壓范圍根據我國2010年-2014年的氣象數據選定為(5670-10550)hPa。在該范圍內,以標稱值1.5μm顆粒為例,計算得到的空氣動力學當量直徑與按公式(1-2)與(1-3)得到的室溫、1個大氣壓下的空氣動力學當量直徑的極差為R=1.62nm。標準物質空氣動力學當量直徑不確定度合成:根據將物理直徑轉化為空氣動力學當量直徑的計算公式(1-1),空氣動力學當量直徑的不確定度計算公式為:但由于根據公式(1-2)和公式(1-3),CDp與CDae也與Dp和Dae相關,因此空氣動力學當量直徑的不確定度不能簡單使用以上公式計算。將公式(1-2)與(1-3)代入公式(1-1)以消去CDp與CDae,得到公式(8-3):則公式(8-3)為描述顆粒密度、物理直徑與空氣動力學當量直徑之間關系的方程,且CDp與CDae之間為非線性關系,并且難以直接推導得到Dae的表達式。參照非線性測量模型不確定度評定相關方法,按照泰勒級數展開,此時不確定度傳播定律為:其中各個偏導數可按隱函數求導法則得到。由前述可以得到空氣動力學當量直徑不確定分量為:a.顆粒的物理尺寸引入的標準不確定度uc(Dp)=16.9nm。b.真密度引入的標準不確定度uc(ρp)=0.0021g/cm3。代入公式(8-4)得到氣溶膠空氣動力學當量直徑定值標準不確定度為:uc(Dae)=17.4nm;考慮到坎寧安滑動修正因子隨溫度與氣壓的變化也可影響公式(8-4)的結果,因此計算了在所設定的氣溫與氣壓范圍內,顆粒的空氣動力學當量直徑定值的標準不確定度,結果表明不確定度在該范圍內基本為一恒定值,因而可使用公式(1-2)與(1-3)得到的室溫、1個大氣壓下的定值標準不確定度作為全范圍內的定值標準不確定度。而對于使用過程中因氣溫與氣壓的變動引入的不確定度,考慮到氣溫與氣壓的變化與測量地點和時間相關,其分布無法預測,故在進行不確定度合成時采用加和的方式進行合成。uc=uc(Dae)+R=19.0nm;則,相對標準不確定度為:同系列其它粒徑定值不確定度分析結果:按照上述相同分析方法,分別計算出其它7種聚苯乙烯顆粒的標準不確定度和擴展不確定度,如表63所示。表63其他7種顆粒空氣動力學當量直徑不確定度的結果定值結果的驗證:采用標準物質對定值方法準確性進行驗證:為了驗證標準物質定值方法的準確性,選用NIST標準物質SRM1962(數均粒徑:(2.982±0.016)μm作為測量對象進行測量,測量方法為:從SRM1962樣品中取1個樣本,然后采用與本發明具體實施方式的定值方法進行測量。通過測量得到SRM1962標準物質的數量平均粒徑為2993nm,該測量結果在標準物質的定值不確定度范圍之內,說明定值方法具有很好的準確性。采用庫爾特粒度測量儀對空氣動力學當量直徑進行驗證:采用美國COULTER公司的MS3型庫爾特粒度儀(精度優于0.5%)對研制的8種聚苯乙烯顆粒空氣動力學當量直徑進行驗證。首先使用Duke標準物質(系列號為4204A和4207A)對MS3型庫爾特粒度儀進行校準,然后再使用MS3型庫爾特粒度儀分別對8種樣品進行測量,每種樣品測量6次,將測量結果的平均值代入公式(1-1)中,計算得到8種標準物質空氣動力學當量直徑,測量及計算結果如表64所示。表64使用庫爾特粒度儀測量結果及空氣動力學當量直徑計算結果由表64可見,將庫爾特粒度分析儀測量結果轉化為空氣動力學當量直徑結果均在空氣動力學定值不確定度范圍內,說明空氣動力學當量直徑的定值可靠。采用單顆粒氣溶膠質譜儀對空氣動力學當量直徑進行測量和驗證:委托廣州禾信公司使用SPAMS0515-R單顆粒氣溶膠質譜儀和掃描電鏡對上述8種顆粒進行空氣動力學當量直徑測量。測量儀器均使用LifeTechnology公司生產的sulfatelatex顆粒(NIST-traceable)進行校準,然后分別對8種顆粒進行測量。每種顆粒均采集至少800個測量數據,計算其平均粒徑。測量結果如表65所示。表65單顆粒氣溶膠質譜儀測量結果平均值空氣動力學標稱直徑μm空氣動力學當量直徑標準值nm測量結果平均值nm1.51491±3815142.01837±4118002.22217±4521772.52505±4925062.82813±5227833.03216±5732353.53748±6437744.04220±714247注:標稱值為3.0μm、3.5μm與4.0μm的顆粒采用掃描電鏡測量。由表65可見,使用氣溶膠質譜儀測量聚苯乙烯顆粒結果均在空氣動力學定值不確定度范圍內,說明空氣動力學當量直徑的定值可靠。采用空氣動力學氣溶膠粒徑譜儀對空氣動力學當量直徑進行測量和驗證:委托北京市環境保護科學研究院使用經校準的空氣動力學氣溶膠粒徑譜儀(APS3321)對上述8種顆粒進行空氣動力學當量直徑測量,測量結果如表66所示。表66使用APS3321測量8種聚苯乙烯顆粒的結果空氣動力學標稱直徑(μm)空氣動力學當量直徑標準值(nm)測量結果平均值(nm)1.51491±3815142.01837±4118292.22217±4522042.52505±4925372.82813±5228413.03216±5731623.53748±6437324.04220±714185由表66可見,使用空氣動力學氣溶膠粒徑譜儀測量聚苯乙烯顆粒結果均在空氣動力學定值不確定度范圍內,說明空氣動力學當量直徑的定值可靠。盡管已用具體實施例來說明和描述了本發明,然而應意識到,以上各實施例僅用以說明本發明的技術方案,而非對其限制;本領域的普通技術人員應當理解:在不背離本發明的精神和范圍的情況下,可以對前述各實施例所記載的技術方案進行修改,或者對其中部分或者全部技術特征進行等同替換;而這些修改或者替換,并不使相應技術方案的本質脫離本發明各實施例技術方案的范圍;因此,這意味著在所附權利要求中包括屬于本發明范圍內的所有這些替換和修改。當前第1頁1 2 3
相關技術
網友詢問留言
已有0條留言
- 還沒有人留言評論。精彩留言會獲得點贊!
1