一種炎癥響應型引導組織再生膜的制備方法與流程
本發明屬于材料表面改性領域,具體涉及一種炎癥響應型釋放抗炎藥物膜的制備方法。
背景技術:
異位骨化(heterotopicossification,ho)是指關節周圍軟組織中出現成熟的板層狀骨的現象。可引起復雜性區域疼痛綜合征、關節僵硬、神經嵌壓等并發癥。迄今為止對于ho的發病機理尚不明晰。但現有研究認為異位骨的形成與軟組織內的間充質干細胞向成骨細胞的不正當分化有關。對于術后異位骨的生成來說,局部炎癥反應引發的局部環境變化是促使這種不正當分化的至關重要的因素【1】。
ho是全人工間盤置換術(tdr)后并發癥中發病率較高的一種。通過對頸椎間盤置換術后ho發生率的meta分析顯示:術后12個月和24個月的合并發病率為44.6%和58.2%,其中重度ho的發病率分別為11.1%和16.7%【2】。較傳統的椎間融合術來說,開展人工間盤置換術的意義核心在于維持脊柱可動性,而間盤假體周圍異位骨化的發生無疑阻礙了這項技術的推廣。如何有效降低人工頸椎間盤置換后異位骨化的發生率是臨床亟待解決的問題。
人工椎間盤置換術后,必定伴有局部炎癥的發生,組織損傷部位的壞死細胞釋放危險信號,固有免疫系統的細胞識別信號,導致一些細胞因子、炎性因子及炎性介質的釋放。這些因子協同作用,誘導間充質干細胞向成骨細胞的不正當分化,從而導致異位骨的生成。其中,轉化生長因子tgf-βs、骨形態發生蛋白bmps及前列腺素2(pge2)被認為在誘導間充質干細胞向成骨細胞分化過程中起關鍵作用。
目前臨床還沒有預防抑制人工頸椎間盤置換術后ho的有效手段,普遍采用局部放射治療和口服非甾體類消炎藥(non-steroidanti-inflammatorydrug,nsaids)的方法。放療的作用機制是通過改變快速分化細胞的dna結構,阻止多能間充質干細胞向成骨細胞的分化,從而抑制ho的發生。然而,放療預防ho局限性很大,容易引發癌變、導致骨不連以及影響生殖能力等。口服nsaids是目前預防術后異位骨化發生所采用的最普遍和最有效的方法。研究表明術前使用nsaids能將術后ho發生率降低約57%-59%。口服nsaids預防術后ho的方法簡單,治療成本相對較低,因此得到了較為廣泛的應用,但其副作用也十分明顯,主要表現為消化道潰瘍,約20-37%的患者因為胃腸道反應而不能完成治療,其次,還會引起肝臟、泌尿和血液系統的不良反應。此外,頸椎間盤置換術后異位骨化的臨床表現最早出現于術后3周,也有術后半年,甚至1年后出現的報道,具有較大的臨床個體差異性。而目前采用的放療或口服nsaids一般都是在圍手術期進行干預,無法針對個體差異性預防抑制。
當炎癥發生的時候,會有大量的巨噬細胞聚集,聚集的巨噬細胞會分泌大量的膽固醇酯酶(cholesterolesterase,ce),并且感染越嚴重釋放的ce量越多,而ce對酰胺鍵及酯鍵具有選擇性酶解作用。將藥物通過酯鍵接枝到膜表面,對炎癥發生過程中產生的酶具有響應性。當炎癥發生時,酯鍵在酶的作用下斷裂,藥物釋放;炎癥程度越高,周圍聚集的巨噬細胞越多,釋放的酶越多,從而釋放的藥物的量就越多。利用ce對酯鍵的選擇性酶解,將藥物分子通過酯鍵固定在膜表面,且連接藥物的酯鍵的含量達到一定數量,就可能構建炎癥響應型釋放藥物的膜。從而達到針對不同患者發病時間及嚴重程度的差異,高效針對性預防抑制炎癥的目的。
參考文獻:
[1]balbonita,gobezier,mamonhj,heterotopicossification:pathophysiology,clinicalfeatures,andtheroleofradiotherapyforprophylaxis.int.j.radiationoncologybiol.phys.,2006,65:1289–1299.
[2]chenj,wangxw,baiws,shenxl,yuanw.prevalenceofheterotopicossificationaftercervicaltotaldiscarthroplasty:ameta-analysis:eurspinej,2012,21:674-680.
技術實現要素:
本申請針對現有預防抑制術后異位骨化發生方法的局限性,提出將臨床常用非甾體類抗炎藥物以酯鍵固定在植入膜材料表面,利用炎癥巨噬細胞釋放的ce對酯鍵的選擇性酶解作用,實現通過炎癥反應的程度控制調節nsaids釋放的目的,從而達到克服目前臨床普遍口服nsaids造成的嚴重副作用,局部高效預防抑制術后異位骨化發生的目的。
1.一種炎癥響應型膜的制備方法,其特征是:高分子膜為基體材料,通過紫外光引發,將itxsp休眠種引入到高分子膜表面;通過紫外光引發,激活itxsp休眠種,在激活表面同時加入引發劑、交聯劑及烯醇類單體,從而在膜表面接枝上一層水凝膠層,改變膜表面的水溶性;將帶羧基的抗炎藥物與烯醇類單體反應,形成含酯基及碳碳雙鍵的單體;最后將該含酯基及碳碳雙鍵的單體、烯醇類單體、交聯劑,在熱引發劑的引發下,在膜表面上再加載上一層含有藥物的凝膠層,其中藥物以酯鍵固定在凝膠層中。
2.根據權利要求1所述的方法,其特征在于,其具體操作步驟為:
(1)將itx丙酮飽和溶液涂覆在高分子膜表面,蓋上石英板,通過紫外引發,將itxsp官能團引入高分子膜表面,得到載有itxsp官能團的聚合物膜;
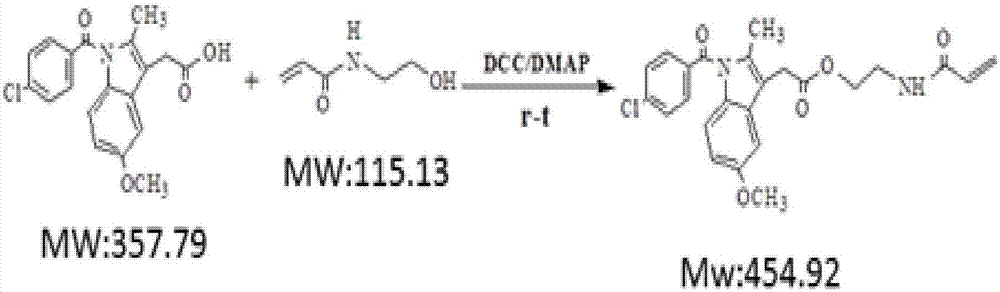
(2)烯醇類單體與帶羧基抗炎藥物在dcc/dmap(n,n'-二環己基碳酰亞胺/4-二甲氨基吡啶)催化下進行酯化反應,從而得到含藥物、酯鍵及雙鍵的單體,將得到的單體記為drug;dcc/dmap即為n,n'-二環己基碳酰亞胺/4-二甲氨基吡啶,其中反應單體與引發劑的投料摩爾比為idcm:hemaa:dcc:dmap=1:3:3:0.5,
(3)將引發劑,交聯劑及含酯基及碳碳雙鍵的單體按照摩爾比=1:1:5,濃度10-30wt%配制成溶液;在載有itxsp官能團的聚合物膜表面,涂覆該溶液,蓋上石英板,通過紫外光引發,從而在該高分子膜表面引入含羥基的凝膠層;
(4)將步驟(2)所得drug、引發劑,交聯劑及乳化劑按照摩爾比6:1:1:0.5配制成質量濃度為10-30wt%的乳液體系.;在含羥基的凝膠層上,涂覆一層該乳液在30℃-70℃烘箱中反應0.5h-2h,獲得加載藥物的凝膠層。
3.進一步,高分子膜厚度為100μm-1mm,具有無孔或有孔的結構,孔徑1-10μm,其制備方法包括:靜電紡絲、熔融澆注或真空模壓法。
4.根據權利要求1所述的方法,其特征在于,高分子膜為聚氨酯及或硅橡膠。
5.進一步,帶羧基抗炎藥為非甾體類抗炎藥物。
6.進一步,烯醇類單體n-羥乙基丙烯酰胺、n-羥甲基丙烯酰胺。
7.進一步,引發劑為過硫酸鉀或過硫酸銨。
8.進一步,交聯劑為n,n’-亞甲基雙丙烯酰胺。
9.進一步,乳化劑為十二烷基磺酸鈉、十二烷基磺酸鈉-明膠的復配體系。
10.進一步,將步驟(1)或步驟(3)紫外光引發的波長為200-400nm,照射時長10-60min,照射距離50mm。
附圖說明
圖1聚氨酯表面加載藥物的過程方法示意圖
其中:
mba:n,n’-亞甲基雙丙烯酰胺交聯劑
heaa:n-羥乙基丙烯酰胺
k2s2o8:過硫酸胺引發劑
sds:十二烷基磺酸鈉表面活性劑
圖2是n-羥乙基丙烯酰胺(hemaa)與吲哚美辛(idcm)在dcc/dmap(n,n'-二環己基碳酰亞胺/4-二甲氨基吡啶)催化下進行酯化反應的方程式:
圖3炎癥響應性藥物釋放膜材料在沒有ce的pbs的緩沖溶液和50u/mlce酶溶液中浸泡3小時后藥物釋放的高效液相色譜曲線
圖4在ce酶中浸泡時間12h-72h后hplc譜圖,可以發現連接c=c的藥物分子的濃度逐漸減少,而吲哚美辛的含量增加,可以看出,酯鍵對膽固醇酯酶具有很高的敏感性。
圖5無脂多糖(lps)刺激(con)以及lps刺激下不同藥物加載量的樣品(y10,y20)與直接加藥品(dm1)對炎癥因子il-6分泌的影響對比
con:空白對照(不刺激,不加藥);lps:脂多糖;y10,y20:材料組(y10:載藥量為10wt%;y20:載藥量為20wt%);dm:直接加入吲哚美辛藥物對照組
具體實施方式
下面通過具體實施例進一步說明本發明,但并不局限于以下的實施例。在閱讀了本發明講授的內容之后,本領域技術人員可以對本發明做各種改動或修改,但凡在本發明的精神和原則之內,這些等價形式的改動、修改等均應包含在本發明的保護范圍之內。
實施例1
(1)在靜電紡絲多孔聚氨酯(pu)表面,將itx丙酮飽和溶液涂覆在聚氨酯表面,將聚氨酯表面全部覆蓋后,蓋上石英板,通過紫外光照射10分鐘(波長200nm,照射距離50mm),將itxsp官能團引入到聚氨酯(pu)膜表面(厚度100μm,平均孔徑1μm)。
(2)通過n-羥乙基丙烯酰胺(hemaa)與吲哚美辛(idcm)在dcc/dmap(n,n′-二環己基碳酰亞胺/4-二甲氨基吡啶)催化下進行酯化反應,從而得到含藥物、酯鍵及雙鍵的單體。其中反應單體與引發劑的投料摩爾比為idcm∶hemaa∶dcc∶dmap=1∶3∶3∶0.5,溶劑為無水dmf,反應溫度為40℃,反應時間24h。先一次性加入除dcc以外其他原料,然后冰浴條件下加入dcc,持續冰浴約20分鐘;反應結束后,冰浴冷卻反應液使n,n′二環己基脲(dcu)析出,抽濾,濾液用水溶液沉淀,靜置/離心至上層液澄清;倒出上層清液,下層沉淀用乙醚超聲脫落,過濾得沉淀,用石油醚清洗3次;以乙酸乙酯和石油醚的混合溶液(體積比6∶1)的流動相過柱子提純,45℃的條件下旋轉獲得產物。收取產物,至于冰箱中保存。通過該方法獲得含c=c的藥物分子。
(3)將過硫酸胺引發劑(k2s2o8),n,n′-亞甲基雙丙烯酰胺交聯劑(mba)以及n-羥乙基丙烯酰胺(heaa)按照摩爾比1∶1∶5配制成溶液,三者質量占溶液總質量的10wt%。在載有itxsp官能團的聚氨酯表面,涂覆配制的溶液,蓋上石英板,通過紫外光引發(波長200nm,時長60min,照射距離50mm),從而在該膜表面引入含羥基的凝膠層。兩次紫外光引發一個有光強一個有距離建議修改為一致
(4)將步驟(2)所得產物、k252o8,mba及sds按照摩爾比6∶1∶1∶0.5配制成質量比為10wt%的乳液體系,將該乳液體系涂覆在步驟(3)獲得的含羥基的凝膠層表面。在70℃烘箱中反應0.5h,在膜表面獲得含藥物的凝膠層,其中藥物以酯鍵固定在凝膠層中。且藥物含量為1.5μg/cm2。1小時藥物釋放達到總量的95%。
實施例2
(1)在靜電紡絲多孔聚氨酯(pu)表面,將itx丙酮飽和溶液涂覆在聚氨酯表面,將聚氨酯表面全部覆蓋后,蓋上石英板,通過紫外光照射10分鐘(波長400nm,照射距離50mm),將itxsp官能團引入到聚氨酯(pu)膜表面(厚度1mm,平均孔徑10μm);
(2)通過n-羥乙基丙烯酰胺(hemaa)與吲哚美辛(idcm)在dcc/dmap(n,n′-二環己基碳酰亞胺/4-二甲氨基吡啶)催化下進行酯化反應,從而得到含藥物、酯鍵及雙鍵的單體。其中反應單體與引發劑的投料摩爾比為idcm∶hemaa∶dcc∶dmap=1∶3∶3∶0.5,溶劑為無水dmf,反應溫度為40℃,反應時間24h。先一次性加入除dcc以外其他原料,然后冰浴條件下加入dcc,持續冰浴約20分鐘;反應結束后,冰浴冷卻反應液使dcu析出,抽濾,濾液用水溶液沉淀,靜置/離心至上層液澄清;倒出上層清液,下層沉淀用乙醚超聲脫落,過濾得沉淀,用石油醚清洗3次;以乙酸乙酯和石油醚的混合溶液(體積比6∶1)的流動相過柱子提純,45℃的條件下旋轉獲得產物。收取產物,至于冰箱中保存。該凝膠層厚度為0.1mm。
(3)將過硫酸胺引發劑(k2s2o8),n,n′-亞甲基雙丙烯酰胺交聯劑(mba)以及n-羥乙基丙烯酰胺(heaa)按照摩爾比1∶1∶5配制成溶液,三者質量占溶液總質量的30wt%。在載有itxsp官能團的聚氨酯表面,涂覆配制的溶液,蓋上石英板,通過紫外光引發(波長400nm,時長30min,照射距離50mm),從而在該膜表面引入合羥基的凝膠層。
(4)將步驟(2)所得產物、k2s2o8,mba及sds按照摩爾比6∶1∶1∶0.5配制成質量比為30wt%的乳液體系,將該乳液體系涂覆在步驟(3)獲得的含羥基的凝膠層表面。在30℃烘箱中反應2h,在膜表面獲得含藥物的凝膠層,其中藥物以酯鍵固定在凝膠層中,且藥物含量為1μg/cm2,1小時藥物釋放達到總量的96%。
實施例3
(1)在靜電紡絲多孔聚氨酯(pu)表面,將itx丙酮飽和溶液涂覆在聚氨酯表面,將聚氨酯表面全部覆蓋后,蓋上石英板,通過紫外光照射10分鐘(波長350nm,照射距離50mm),將itxsp官能團引入到聚氨酯(pu)膜表面(厚度0.5mm,平均孔徑6μm);
(2)通過n-羥乙基丙烯酰胺(hemaa)與吲哚美辛(idcm)在dcc/dmap(n,n′-二環己基碳酰亞胺/4-二甲氨基吡啶)催化下進行酯化反應,從而得到含藥物、酯鍵及雙鍵的單體。其中反應單體與引發劑的投料摩爾比為idcm∶hemaa∶dcc∶dmap=1∶3∶3∶0.5,溶劑為無水dmf,反應溫度為40℃,反應時間24h。先一次性加入除dcc以外其他原料,然后冰浴條件下加入dcc,持續冰浴約20分鐘;反應結束后,冰浴冷卻反應液使dcu析出,抽濾,濾液用水溶液沉淀,靜置/離心至上層液澄清;倒出上層清液,下層沉淀用乙醚超聲脫落,過濾得沉淀,用石油醚清洗3次;以乙酸乙酯和石油醚的混合溶液(體積比6∶1)的流動相過柱子提純,45℃的條件下旋轉獲得產物。收取產物,至于冰箱中保存。該凝膠層厚度為0.1mm。
(3)將過硫酸胺引發劑(k2s2o8),n,n′-亞甲基雙丙烯酰胺交聯劑(mba)以及n-羥乙基丙烯酰胺(heaa)按照摩爾比1∶1∶5配制成溶液,三者質量占溶液總質量的20wt%。在載有itxsp官能團的聚氨酯表面,涂覆配制的溶液,蓋上石英板,通過紫外光引發(波長350nm,時長45min,照射距離50mm),從而在該膜表面引入合羥基的凝膠層。
(4)將步驟(2)所得產物、k252o8,mba及sds按照摩爾比6∶1∶1∶0.5配制成質量比為20wt%的乳液體系,將該乳液體系涂覆在步驟(3)獲得的合羥基的凝膠層表面。在40℃烘箱中反應1.5h,從而將藥物以酯鍵固定在靜電紡絲型聚氨酯膜表面。在膜表面獲得合藥物的凝膠層,其中藥物以酯鍵固定在凝膠層中,且藥物含量為0.8μg/cm2.1小時藥物釋放達到總量的98%。
實施例4
將實施例3中的電紡聚氨酯pu膜替換成熔融澆注法制備的無孔pu膜,其他實驗條件與實施例3相同。將藥物以酯鍵固定在靜電紡絲型聚氨酯膜表面的加載藥物的凝膠層中,其中藥物含量為0.3μg/cm2。1小時藥物釋放達到總量的97%。
實施例5
將實施例1中的電紡聚氨酯pu膜替換成真空模壓法制備的無孔pu膜,其他實驗條件與實施例1相同。將藥物以酯鍵固定在靜電紡絲型聚氨酯膜表面加載藥物的凝膠層中,其中藥物含量為0.6μg/cm2.1小時藥物釋放達到總量的98%。
實施例6
將實施例3中的電紡聚氨酯pu膜替換成熔融澆注法制備的0.5mm厚的無孔硅橡膠膜,其他實驗條件與實施例3相同。將藥物以酯鍵固定在熔融澆注法制備的無孔硅橡膠膜表面的加載藥物的凝膠層中,其中藥物含量為0.3μg/cm2。1小時藥物釋放達到總量的97%。
實施例7
將實施例1中的n-羥乙基丙烯酰胺(hemaa)替換成n-羥甲基丙烯酰胺(ham),其他實驗條件與實施例1相同。n-羥甲基丙烯酰胺(ham)與吲哚美辛(idcm)合成的產物通過質譜和核磁測試,發現在質譜圖中出現分子量為440.9的物質,且對比核磁圖中相應的h的化學位移,可以確定獲得所需產物。獲得將藥物以酯鍵固定在靜電紡絲型聚氨酯膜表面厚的加載藥物的凝膠層中,其中藥物含量為1.3μg/cm2。1小時藥物釋放達到總量的97%。
實施例8
將實施例1中的抗炎藥物吲哚美辛替換成阿司匹林,其他實驗條件與實施例1相同。測定阿司匹林與n-羥乙基丙烯酰胺(hemaa)反應產物的質譜圖和核磁圖,發現在質譜圖中出現分子量為277.29的物質,且對比核磁圖中相應的h的化學位移,可以確定獲得所需產物。獲得將藥物以酯鍵固定在靜電紡絲型聚氨酯膜表面加載藥物的凝膠層中,其中藥物含量為1.2μg/cm2。1小時藥物釋放達到總量的96%。
實施例9
將實施例1中的熱引發劑過硫酸鉀替換成過硫酸銨,其他實驗條件與實施例1相同。獲得將藥物以酯鍵固定在靜電紡絲型聚氨酯膜表面加載藥物的凝膠層中,其中藥物含量為1.3μg/cm2。1小時藥物釋放達到總量的98%。
實施例10
將實施例1中的乳化劑十二烷基磺酸鈉換成十二烷基磺酸鈉-明膠的復配體系,其他實驗條件與實施例1相同。獲得將藥物以酯鍵固定在靜電紡絲型聚氨酯膜表面厚的加載藥物的凝膠層中,其中藥物含量為1.5μg/cm2。1小時藥物釋放達到總量的94%。
- 還沒有人留言評論。精彩留言會獲得點贊!