提高金屬粉在高黏體系中分散穩定性的顆粒表面處理方法與流程
本發明涉及一種顆粒表面處理方法,具體涉及一種提高金屬粉在高黏體系中分散穩定性的顆粒表面方法,屬于材料表面改性技術領域。
背景技術:
金屬粉由于其密度較高,作為填料添加到高黏體系中分散穩定性不好,容易發生沉降,導致復合材料組分、密度均勻性受到影響,最終給材料的性能帶來不良影響。
對于高黏懸浮體系的分散穩定性研究,目前最常用和最可靠的方法是沉降法。根據斯托克斯定律,懸浮液中固態顆粒在重力場作用下的沉降速率主要與顆粒粒徑、顆粒與液體的密度差及體系黏度有關。近年來研究發現,采用一些高分子聚合物來對金屬粉進行表面處理也有很好的分散穩定效果,如聚丙烯酰胺、聚丙烯酸鈉、聚乙二醇等,但在混合過程中這些聚合物包覆層會出現不同程度的脫落,導致分散效果不佳。
自然界中貽貝分泌的蛋白質具有強的黏附特性,研究者通過研究發現其中發揮黏附作用的關鍵物質是粘附蛋白中含有的兒茶酚類物質,這類物質在一定環境條件下可發生自聚反應,在多種材料表面形成緊密的包覆層。因此,利用這種簡單的自聚反應可在金屬粉表面形成一層薄且牢固的包覆層,一方面使聚合物包覆層在混合過程中不易脫粘,另一方面兒茶酚物質中豐富的活性基團可提高其與流動相的相互作用力,進一步提高金屬粉在高黏懸浮體系中的分散穩定性。
技術實現要素:
本發明的目的在于解決金屬粉在高黏體系中分散穩定性不好的問題,提供了一種簡易的金屬粉表面包覆方法,減緩了其沉降速率,提高金屬粉在高黏體系中分散穩定性。且本發明使用的方法處理條件溫和、試劑環保無毒,后處理過程簡便。
本發明采用的技術方案如下:
一種提高金屬粉在高黏體系中分散穩定性的顆粒表面處理方法,包括以下步驟:
步驟一、將金屬粉顆粒超聲分散在乙醇中制得濃度為1mg/ml~100mg/ml,繼續超聲清洗1min~20min;
步驟二、將清洗后的金屬粉顆粒進行抽濾后,置于20℃~120℃的烘箱中干燥0.5h~12h;
步驟三、配置pH為8~14的堿性溶液;
步驟四、在堿性溶液中溶解兒茶酚類物質,溶解后兒茶酚類物質的濃度為0.1g/L~20g/L,完全溶解后溶液pH值仍大于7;
步驟五、將金屬粉置于上述配置的溶液中,使用攪拌器以100r/min~1000r/min的轉速攪拌,使金屬粉完全分散在溶液中形成分散液,攪拌時間為0.5h~24h;
步驟六、將攪拌完成的分散液進行抽濾清洗,使用蒸餾水清洗2~5遍;
步驟七、將清洗后的材料置于50℃~80℃烘箱中1h~24h,得到表面包覆的金屬粉。
更進一步的方案是:所述金屬粉包括:鋁粉、鐵粉、銅粉、鋅粉中的任意一種,所述金屬粉的粒徑范圍為:0.01μm~100μm。
更進一步的方案是:所述兒茶酚類物質為多巴胺。
更進一步的方案是:所述高黏體系,流動相為:端羥基聚丁二烯(HTPB)、季戊四醇丙烯醛樹酯(123樹酯)或醋酸丁酸纖維素(CAB)。
更進一步的方案是:所述步驟三中堿性溶液為三羥甲基氨基甲烷(Tris)堿性緩沖溶液、碳酸氫鈉弱堿性溶液、氨水、氫氧化鈉或氫氧化鉀等強堿性溶液中的一種。
本發明的優點:(1)本發明金屬粉表面處理工藝簡單,只需在配置的溶液中攪拌即可,可大規模批量制備;(2)本發明金屬粉表面處理材料均勻性和完整性良好,不會出現局部包覆的情況,使材料的均勻性得到顯著提升;(3)本發明兒茶酚類物質與金屬粉表面的粘附力強,不會在混合過程中出現脫落和聚集現象,可提高顆粒在粘結劑中的分散穩定性。
附圖說明
圖1為實施例1中鋁粉表面處理前后的SEM照片。
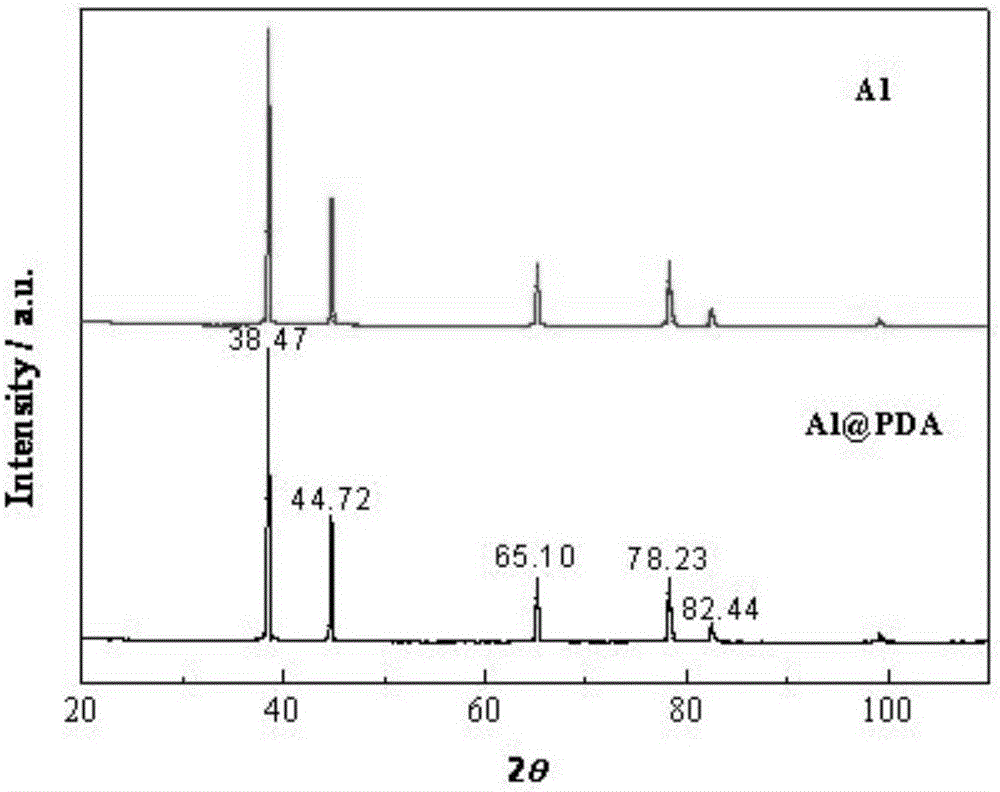
圖2為實施例2中鋁粉表面處理前后的XRD測試數據。
圖3為實施例4中制備的表面處理后鋁粉的XPS測試數據。
圖4為實施例1中處理后的鋁粉與未經處理鋁粉分別與HTPB充分混勻并靜置24h后的沉降程度對比。
具體實施方式
為了更清楚的說明本申請文件實施例或現有技術中的技術方案,下面結合實施例對本發明做詳細描述。
實施例1
一種提高金屬粉在高黏體系中分散穩定性的顆粒表面處理方法,包括以下步驟:
(1)將粒徑為5μm的鋁粉超聲分散在乙醇中制得濃度為5mg/ml,超聲清洗10min;
(2)將清洗后的固體顆粒進行抽濾后,置于60℃的烘箱中干燥5h;
(3)配置10mM濃度的三羥甲基氨基甲烷的緩沖水溶液100ml;
(4)在Tris溶液中溶解鹽酸多巴胺,溶解后鹽酸多巴胺的濃度為2g/L,溶液pH值為8.5;
(5)將6g鋁粉置于上述配置的溶液中,使用攪拌器以400r/min的轉速攪拌混合分散液,攪拌時間為12h;
(6)將攪拌完成的分散液進行抽濾清洗,使用蒸餾水清洗3遍;
(7)將清洗后的材料置于80℃烘箱中干燥6h,得到表面處理后的鋁粉。
實施例2
一種提高金屬粉在高黏體系中分散穩定性的顆粒表面處理方法,包括以下步驟:
(1)將粒徑為50μm的銅粉超聲分散在乙醇中制得濃度為5mg/ml,超聲清洗10min;
(2)將清洗后的固體顆粒進行抽濾后,置于60℃的烘箱中干燥5h;
(3)配置10mM濃度的三羥甲基氨基甲烷的緩沖水溶液100ml,
(4)在Tris溶液中溶解鹽酸多巴胺,溶解后鹽酸多巴胺的濃度為4g/L,完全溶解后溶液pH值為8.5;
(5)將6g銅粉置于上述配置的溶液中,使用攪拌器以400r/min的轉速攪拌混合分散液,攪拌時間為12h;
(6)將攪拌完成的分散液進行抽濾清洗,使用蒸餾水清洗3遍;
(7)將清洗后的材料置于80℃烘箱中干燥6h,得到表面處理后的銅粉。
實施例3
一種提高金屬粉在高黏體系中分散穩定性的顆粒表面處理方法,包括以下步驟:
(1)將粒徑為0.5μm的鐵粉超聲分散在乙醇中制得濃度為5mg/ml,超聲清洗10min;
(2)將清洗后的固體顆粒進行抽濾后,置于60℃的烘箱中干燥5h;
(3)配置10mM濃度的三羥甲基氨基甲烷的緩沖水溶液100ml;
(4)在Tris溶液中溶解鹽酸多巴胺,溶解后鹽酸多巴胺的濃度為3g/L,完全溶解后溶液pH值為8.5;
(5)將3g鋁粉置于上述配置的溶液中,使用攪拌器以400r/min的轉速攪拌混合分散液,攪拌時間為18h;
(6)將攪拌完成的分散液進行抽濾清洗,使用蒸餾水清洗3遍;
(7)將清洗后的材料置于60℃烘箱中干燥8h,得到表面處理后的鐵粉。
實施例4
一種提高金屬粉在高黏體系中分散穩定性的顆粒表面處理方法,包括以下步驟:
(1)將粒徑為50μm的鋅粉超聲分散在乙醇中制得濃度為5mg/ml,超聲清洗10min;
(2)將清洗后的固體顆粒進行抽濾后,置于60℃的烘箱中干燥5h;
(3)配置濃度為10mL/L的氨水溶液100mL;
(4)在氨水溶液中溶解鹽酸多巴胺,溶解后鹽酸多巴胺的濃度為2g/L,完全溶解后溶液pH值為10;
(5)將6g鋁粉置于上述配置的溶液中,使用攪拌器以400r/min的轉速攪拌混合分散液,攪拌時間為10h;
(6)將攪拌完成的分散液進行抽濾清洗,使用蒸餾水清洗3遍;
(7)將清洗后的材料置于80℃烘箱中干燥6h,得到表面處理后的鋅粉。
實施例5
一種提高金屬粉在高黏體系中分散穩定性的顆粒表面處理方法,包括以下步驟:
(1)將粒徑為50μm的鋁粉超聲分散在乙醇中制得濃度為5mg/ml,超聲清洗10min;
(2)將清洗后的固體顆粒進行抽濾后,置于60℃的烘箱中干燥5h;
(3)配置濃度為5g/L的氫氧化鈉溶液100mL;
(4)在氫氧化鈉溶液中溶解鹽酸多巴胺,溶解后鹽酸多巴胺的濃度為4g/L,完全溶解后溶液pH值為11;
(5)將6g鋁粉置于上述配置的溶液中,使用攪拌器以400r/min的轉速攪拌混合分散液,攪拌時間為12h;
(6)將攪拌完成的分散液進行抽濾清洗,使用蒸餾水清洗3遍;
(7)將清洗后的材料置于80℃烘箱中干燥6h,得到表面處理后的鋁粉。
根據實施例,結合附圖進行分析。其中附圖1為實施例1中鋁粉表面處理前后的SEM照片。其中圖1的右圖為處理后的照片。由圖可以看出,處理后的鋁粉表面沉積了一層均勻而致密的薄膜,包覆效果較好。附圖2為實施例2中鋁粉表面處理前后的XRD測試數據,通過測試數據可以看出,包覆前后出峰位置一致,說明本發明的表面處理方法不會對鋁粉的晶型產生影響,且包覆物聚多巴胺為無定形態。附圖3為實施例4中制備的表面處理后鋁粉的XPS測試數據。通過測試數據可看出,改性鋁粉表面含有大量的C、N和O元素,且遠遠多于Al元素,證明聚多巴胺對鋁粉的包覆效果較好。附圖4為實施例1中處理后的鋁粉與未經處理鋁粉分別與HTPB充分混勻并靜置24h后的沉降程度對比。由圖可見懸浮體系靜置24h后,未處理的鋁粉已經由于沉降而分層,懸浮體系上端出現清液,而經過包覆處理的樣品在HTPB中分散依然很均勻,整個懸浮體系顏色均一,表明表面處理后的鋁粉在HTPB中的分散穩定性要明顯優于未處理的鋁粉。
盡管這里參照本發明的解釋性實施例對本發明進行了描述,上述實施例僅為本發明較佳的實施方式,本發明的實施方式并不受上述實施例的限制,應該理解,本領域技術人員可以設計出很多其他的修改和實施方式,這些修改和實施方式將落在本申請公開的原則范圍和精神之內。
- 還沒有人留言評論。精彩留言會獲得點贊!