一種特異靶向人ABCG2基因的sgRNA導向序列及其應用的制作方法
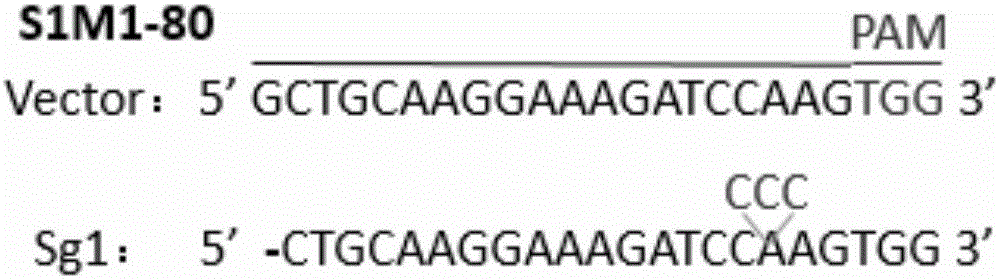
本發明屬于基因工程應用領域,尤其涉及一種特異靶向人ABCG2基因的sgRNA導向序列及其應用。
背景技術:
:CRISPR-Cas9是細菌和古細菌在進化過程中演變出來的一種用于抵御外來侵害的免疫入侵系統,包括抵御入侵的病毒以及外源DNA。在現代基因工程應用領域與TALEN(transcriptionactivator-likeeffectornuclease)以及ZFN(zinc-fingernuclease)技術并列成為三大基因組編輯工具。相比較于TALEN及ZFN技術,CRISPR-Cas9技術具有特異性DNA識別能力,在第二類CRISPR系統中,Cas9核酸內切酶在sgRNA引導下切割雙鏈DNA,造成基因組雙鏈斷裂,利用細胞基因組修復的不穩定性產生非特異重組來產生修復錯誤(插入或者缺失),從而可能產生移碼突變而造成基因功能的喪失,實現基因敲除的目的。其sgRNA的設計和合成工作量遠遠小于TALEN和ZFN識別模塊的構建過程,且毒性遠遠低于ZFN技術。但是CRISPR-Cas9技術也有上下文依賴性的缺點,目前只能應用于上游有PAM序列的靶位點。多藥耐藥性(MDR)是指對一種藥物具有耐藥性的同時,對其他結構不同,作用靶點不同的抗腫瘤藥物也具有耐藥性。多藥耐藥性是導致癌癥藥物治療和腫瘤化療失敗的重要原因之一。而多藥耐藥性的主要機制之一就是ABC(ATP-bindingcassette)轉運蛋白的過量表達,它們可由ATP水解供能來將抗癌藥物泵出細胞外。它主要的家族成員包括ABCB1(P-GP)、ABCC1(MRP1)和ABCG2(BCRP)等。細胞中ABCG2的高表達將會導致多藥耐藥的發生從而使治療失敗,因此抑制或消除ABCG2的表達能夠有效的解決在腫瘤治療中出現的多藥耐藥問題。技術實現要素:為了克服某些高表達ABCG2蛋白的細胞出現多耐藥的缺點與不足,本發明的首要目的在于提供一種特異靶向人ABCG2基因的sgRNA導向序列。該sgRNA導向序列可以用于敲除人ABCG2基因,進而抑制或消除ABCG2的表達。本發明的另一目的在于提供一種利用CRISPR-Cas9系統敲除人ABCG2基因的方法。該方法采用CRISPR-Cas9技術對ABCG2基因進行編輯,使其正常序列發生缺失或者突變從而達到敲除該基因的目的。本發明的再一目的在于提供上述特異靶向人ABCG2基因的sgRNA導向序列的應用。本發明的目的通過下述技術方案實現:一種特異靶向人ABCG2基因的sgRNA導向序列,為Sg1;其核苷酸序列為:Sg1:5'-GCTGCAAGGAAAGATCCAAG-3',位于基因ABCG2第三個外顯子;一種利用CRISPR-Cas9系統敲除人ABCG2基因的方法,包含如下步驟:(1)在上述導向序列的5'端加上CACCG得到正向寡核苷酸;同時根據導向序列獲得其對應的DNA互補鏈,并且在其5'端加上AAAC和其3'端加上C得到反向寡核苷酸;分別合成上述正向寡核苷酸和反向寡核苷酸,將合成的正向寡核苷酸和反向寡核苷酸變性,退火,形成雙鏈;(2)將步驟(1)制得的雙鏈與Cas9載體連接,得到重組敲除表達載體;(3)將步驟(2)制得的重組敲除表達載體與包裝系統共轉染包裝細胞,然后收獲病毒純化并濃縮,得到病毒顆粒;(4)將步驟(3)制得的病毒顆粒感染細胞,篩選穩轉細胞,得到成功敲除ABCG2基因的細胞。步驟(2)中所述的Cas9載體優選為lentiCRISPRv2載體;步驟(3)中所述的包裝細胞優選為293T細胞;步驟(3)中所述的包裝系統中的包裝載體優選為pMD2.G和psPAX2;步驟(4)中所述的細胞優選為腫瘤多藥耐藥性細胞;步驟(4)中所述的細胞進一步優選為S1M1-80;所述的特異靶向人ABCG2基因的sgRNA導向序列在制備抗腫瘤多藥耐藥性藥物中的應用;所述的腫瘤優選為人結直腸癌;本發明相對于現有技術,具有如下的優點及效果:(1)本發明根據sgRNA導向序列設計合成兩條單鏈oligo序列,退火形成雙鏈,然后與Cas9載體連接,利用Cas9載體將sgRNA以及CRISPR系統引入目標細胞中,Cas9蛋白會在sgRNA的引導下找到與其匹配的DNA序列,進行剪切,實現ABCG2基因的敲除。(2)載體中含有Puromycin抗性基因,利用Puromycin對細胞進行篩選,未轉入lentiCRISPRv2載體的細胞將在篩選過程中被淘汰。(3)本發明提供的特異靶向人ABCG2基因的sgRNA導向序列,可通過CRISPR-Cas9系統敲除或編輯ABCG2基因,進而抑制或消除ABCG2的表達能夠有效的解決在腫瘤治療中出現的多藥耐藥問題。附圖說明圖1是特異靶向人ABCG2基因的sgRNA導向序列的序列及其位置示意圖。圖2是應用本發明設計的sgRNA對高表達ABCG2的細胞株進行編輯后抽提細胞mRNA逆轉錄成cDNA后PCR而后進行測序與成功轉染空載lentiCRISPRv2的細胞株進行對比的測序分析圖。圖3是實施例2PCR產物測序結果的測序峰圖。圖4是成功敲除ABCG2基因的細胞株的ABCG2蛋白表達量的westernblot結果分析圖。圖5是Cisplatin、Doxorubicin和Mitoxantrone處理后,成功敲除ABCG2基因的細胞株的細胞活性結果分析圖。圖6是應用本發明設計的sgRNA對高表達ABCG2的細胞株進行編輯后的藥物積累實驗結果結果分析圖。圖7是實施例3藥物積累實驗的流式結果分析圖。圖8是實施例3藥物積累實驗流式結果的量化處理分析圖。具體實施方式下面結合實施例及附圖對本發明作進一步詳細的描述,但本發明的實施方式不限于此。實施例中所使用的細胞株均購自ATCC,lentiCRISPRv2載體購自Addgene,內切酶BsmBⅠ購自Biolabs,Polyetherimide(PEI)購自Ploysciences,Puromycin和Polybreen購自Sigma。實施例1(1)sgRNA設計根據人ABCG2基因的基因組序列(geneID:9429),設計1個靶向人ABCG2基因的sgRNA。20nt的寡核苷酸sgRNA導向序列為:Sg1:5'-GCTGCAAGGAAAGATCCAAG-3'(位于基因ABCG2第三個外顯子)(圖1);在其5'端加上CACCG得到正向寡核苷酸(Forwardoligo)(見表1中Sg1-F);根據導向序列獲得其對應的DNA互補鏈,并且在其5'端加上AAAC和其3'端加上C得到反向寡核苷酸(Reverseoligo)(見表1中Sg1-R)。分別合成上述正向寡核苷酸和反向寡核苷酸,將合成的sgRNA寡聚核苷酸的Forwardoligo和Reverseoligo成對變性,退火;退火后可以形成連入表達載體lentiCRISPRv2載體的雙鏈,同時將設計好的gRNA靶點序列進行blast比對以排除非特異性的靶切位點,具體寡核苷酸序列見表1。表1sgRNA導向序列的寡核苷酸序列Sequence(5'to3')Sg1-FCACCGGCTGCAAGGAAAGATCCAAGSg1-RAAACCTTGGATCTTTCCTTGCAGCC(2)構建表達sgRNA的載體病毒載體lentiCRISPRv2載體有BsmBⅠ酶切位點,用BsmBⅠ酶切,其中,酶切體系(20μL體系)為:BsmBⅠ1μL;10×NEbuffer2μL;質粒1μL;ddH2O16μL;酶切條件為:37℃酶切1h;將酶切后的載體lentiCRISPRv2分別與步驟(1)制得的退火雙鏈利用T4連接酶進行連接,連接體系(10μL)為:退火雙鏈(Sg1)2μL,lentiCRISPRv2載體2μL,10×NEBT4DNALigaseBuffer1μL,T4DNALigase1μL,ddH2O4μL;連接條件為:16℃連接過夜;將連接產物轉化感受態細胞stbl3,具體轉化方法為:-80℃取出感受態細胞stbl3,與冰上溶解;然后50μL感受態細胞中加入1μL的上述連接產物,混勻后冰浴30min;42℃水浴90s,過程中勿搖動;冰上冷卻1~2min;然后加入800μLLB培養基,37℃搖床30min;涂AMP+板(100μg/mL)培養過夜,挑取陽性克隆后37℃搖床過夜進行擴大培養,并用HipurePlasmidMicroKitC(Magen)抽提質粒并測序驗證,得到表達sgRNA的載體lentiCRISPRv2-hABCG2-Sg1(導向序列為Sg1)(hABCG2表示為人基因ABCG2)。實施例2(1)核心質粒與包裝質粒pMD2.G和psPAX2共轉染至293T細胞中培養293T細胞,待293T細胞的匯集率達到50%~60%,種板后12~18h為最佳轉染時間;轉染前更換新鮮培養液,60mm小皿中加入3mL培養基;轉染時質粒的使用量為核心質粒(實施例1制得的lentiCRISPRv2-hABCG2-Sg1,另取lentiCRISPRv2空載體作為對照)4μg、psPAX23μg、pMD2.G1μg、PEI24μL,補充DMEM至總體積為200μL,加入順序分別為DMEM、PEI和質粒DNA(核心質粒、pMD2.G和psPAX2);然后室溫靜止30min,使PEI與質粒DNA充分聚合,聚合結束后將轉染體系逐滴加入上述培養有293T細胞的小皿中,輕輕搖勻后放入37℃、5%CO2培養箱中繼續培養;(2)病毒的收獲與濃縮收集轉染后24h、48h、72h、96h293T細胞的上清液,每次收取病毒后再補充3mL新鮮培養液于小皿中,先收獲的病毒原液用封口膜封閉后可暫存4℃冰箱中;待病毒液全部收集完成后1000rpm離心5min,以去除細胞碎片,用0.45μm濾膜過濾去除細胞碎片及其他雜質;取過濾后的病毒原液于100kD超濾柱內,4℃,4000g離心30min,收集濾膜內剩余約300μL病毒濃縮液,50μL每管分裝于1.5mLEP管內,置-80℃可長期保存,避免反復凍融;(3)包裝病毒顆粒分別感染目的細胞株S1M1-806孔板內提前一天種好待感染的目的細胞株S1M1-80(人結直腸癌多藥耐藥細胞),細胞感染時細胞匯集率達到40~50%為宜;感染前用1mL新鮮培養基換液,用另外1mL新鮮培養基稀釋步驟(2)制得的50μL病毒濃縮液,加入2μL聚凝胺(Polybreen,10mg/mL,最終工作濃度為10μg/mL)混勻后,逐滴均勻滴入六孔板1個孔內,輕輕晃勻;依據不同細胞株的性質,于感染后6~48h換新鮮培養液;慢病毒介導的基因會在48~96h期間陸續表達,如果效率不理想可以進行反復感染,被感染表達目的基因的細胞理論上均為穩定株;核心載體有Puro(嘌呤霉素)的篩選標記,可以利用Puromycin進行穩定株的篩選;篩選之前測定實驗所用細胞株Puromycin的全部致死濃度為30μg/mL,用全部致死濃度對感染后細胞S1M1-80進行篩選;(4)穩定感染細胞株的篩選與培養利用30μg/mL的Puromicin對感染后的細胞株S1M1-80進行篩選,篩選持續20d,對篩選后的細胞進行培養;(5)根據所設計的兩段sgRNA序列設計鑒定引物,用于對敲除后目的片段進行鑒定,所設計引物如表2所示:表2鑒定引物序列Sequence(5'to3')Detection1-FGACCTGAAGGCATTTACTGAAGGAGCDetection1-RGCTCCATTCCGATCGAATCCCTTTTTCTTTC利用HiPureTotalRNAMiniKit對穩定感染細胞株S1M1-80進行mRNA提取,利用StarScriptⅡFirst-strandcDNASynthesisMix逆轉錄成cDNA,并利用鑒定引物進行PCR鑒定。其中,對含有lentiCRISPRv2-hABCG2-Sg1的病毒顆粒轉染后的細胞株S1M1-80,其逆轉錄體系(20μL)為:RNA模板7μL;Oligo(dT)18(50μM)1μL;2×ReactionMix10μL;StarscripⅡRTMix1μL;DEPC-H2O2μL;逆轉錄條件:42℃孵育50min,85℃加熱5min失活ScriptⅡ。對上述cDNA進行PCR,其反應體系(50μL):10×buffer5μL;dNTP1μL;模板DNA1μL;Detection1-F(10μM)1μL;Detection1-R(10μM)1μL;Pfu高保真酶1μL;ddH2O40μL;PCR反應擴增條件:95℃初始變性4min;95℃變性30S,56℃退火30S,72℃延伸2min,35個循環;72℃最后延伸10min;結果如圖2和圖3所示,表明:測序結果表明編輯組發生了缺失和插入突變,對照組沒有發生變化。本發明發成功建立了敲除ABCG2基因的細胞株S1M1-80sg1。實施例3體外細胞實驗對篩選后細胞進行鑒定人基因ABCG2敲除效果(1)westernblot利用westernblot實驗鑒定實施例2中成功敲除ABCG2基因的細胞株S1M1-80sg1、的ABCG2蛋白表達量,成功轉染空載體lentiCRISPRv2的細胞株S1M1-80作為空白對照,S1作為陽性對照,S1M1-80作為陰性對照;其中,一抗為Anti-ABCG2(MMSC-58222,購自santacruze),二抗為Anti-mouseIgG,HRP-linkedAntibody(貨號7076,cellsignaling),具體方法為常規westernblot操作流程。結果如圖4所示,在S1M1-80細胞中,轉入sg1的細胞與只轉入載體的細胞ABCG2表達量有明顯差異,sg1中幾乎未表達ABCG2蛋白,可作為ABCG2基因被敲除的證據之一。(2)MTT法檢測細胞活性:選取ABCG2的特異性底物Mitoxantrone(購自LC)和Doxorubicin(購自LC)與非特異性底物cisplatin(購自LC)用MTT法檢測檢測成功敲除ABCG2基因的細胞株S1M1-80sg1對上述藥物的敏感性。將細胞S1M1-80sg1及成功轉染空載lentiCRISPRv2的細胞株S1M1-80對照細胞以每孔3000~5000個細胞的數量接種到96孔板,待細胞貼壁后,分別加入不同濃度的Mitoxantrone(1、3、10、30、100、300μM)、Doxorubicin(1、3、10、30、100μM)、cisplatin(1、3、10、30、100、300μM)。培養72h后,每孔加入10μL5mg/mL的MTT再培養4h,然后棄培養液每孔加入100μL的DMSO,用酶標儀在570nm讀取每孔的吸光值。用Bliss法計算半數抑制濃度值(IC50)。結果如圖5所示,與單獨轉入載體的對照細胞相比,成功敲除ABCG2基因的細胞株(S1M1-80)對Mitoxantrone和Doxorubicin的敏感性增加,IC50值明顯降低。而對于ABCG2的非特異性底物Cisplatin來說,對照組與成功敲除ABCG2基因的細胞株對比,IC50值并沒有差異。這可作為利用CRISPR-Cas9系統對S1M1-80的ABCG2基因進行敲除成功的證據之一。(3)藥物積累實驗基因ABCB2編碼的高度糖基化跨膜蛋白ABCG2,它通過消耗ATP而將藥物從細胞內泵出,從而降低腫瘤細胞內的藥物濃度。選擇3種ABCG2的底物:Mitoxantrone、Doxorubicin和Rh-123(購自sigma-Aldrich)流式觀察這三種分子在細胞內的積累情況:將細胞S1M1-80sg1及成功轉染空載lentiCRISPRv2的細胞株S1M1-80對照細胞以每孔2.5×105個的數量接種到6孔板,待細胞貼壁后,分別加入10μM的Mitoxantrone、Doxorubicin和Rh-123(Rhodamine123),培養2h后,PBS潤洗3遍,然后將細胞用胰酶消化,用PBS重懸后用流式細胞分析儀檢測熒光強度,然后用Flowjo軟件進行定量分析;另外,選擇3種ABCG2的底物:Mitoxantrone、Doxorubicin和Rh-123(購自sigma-Aldrich)共聚焦20倍鏡觀察這三種分子在細胞內的積累情況:先在6孔板里滴入少量培養基,然后放無菌蓋玻片(22×22mm),將細胞S1M1-80sg1及成功轉染空載lentiCRISPRv2的細胞株S1M1-80對照細胞以每孔2.5×105個的數量接種到6孔板,待細胞貼壁后,分別加入10μM的Mitoxantrone、Doxorubicin和Rh-123(Rhodamine123),培養2h后,吸掉培養基,用PBS清洗2次。用4%的多聚甲醛(溶劑為PBS)室溫固定10min,吸掉多聚甲醛.加入PBS在搖床上快搖10min,然后加入新的PBS再快搖10min,如此操作2次,清洗掉殘余的多聚甲醛。取200μLDapi溶液100nM對細胞進行染色,避光室溫孵育15min。用PBS清洗蓋玻片后置于載玻片(25×75mm)上,封片而后于共聚焦顯微鏡20倍鏡下觀察。結果如圖6、7、8所示,編輯組(S1M1-80sg1)與對照組相比熒光強度明顯增多,證明編輯組細胞的ABCG2沒有發揮功能將三種藥物泵出細胞。而對照組的ABCG2能正常的發揮藥泵功能,因此熒光強度較低。表明編輯組ABCG2基因被成功敲除。上述實施例為本發明較佳的實施方式,但本發明的實施方式并不受上述實施例的限制,其他的任何未背離本發明的精神實質與原理下所作的改變、修飾、替代、組合、簡化,均應為等效的置換方式,都包含在本發明的保護范圍之內。當前第1頁1 2 3
相關技術
網友詢問留言
已有0條留言
- 還沒有人留言評論。精彩留言會獲得點贊!
1
- 采用寡核苷酸介導的基因修復提高靶向基因修飾的效率的方法和組合物與流程
- CRISPR/Cas9靶向敲除人ezrin基因增強子關鍵區的方法及其特異性gRNA與流程
- CRISPR/Cas9 靶向敲除人PD?1基因及其特異性gRNA的制作方法與工藝
- 靶向人TSPAN8基因的siRNA及其應用的制作方法與工藝
- 靶向人TSPAN8基因的siRNA及其應用的制作方法與工藝
- CRISPR/Cas9靶向敲除人乙肝病毒P基因及其特異性gRNA的制作方法與工藝
- 一種特異靶向人ABCG2基因的sgRNA導向序列及其應用的制作方法與工藝
- 一種利用CRISPR?Cas9靶向敲除MSTN基因的方法與流程
- CRISPR/Cas9靶向敲除人CNE10基因及其特異性gRNA的制作方法與工藝
- 用于豬APN基因編輯的靶向sgRNA、修飾載體及其制備方法和應用與流程