一種不對稱共軛二炔的合成方法與流程
本發明屬于有機合成技術領域,具體涉及一種不對稱共軛二炔的合成方法。
背景技術:
共軛二炔烴是有機合成領域的重要原料和中間體,可以作為起始原料用于合成大環輪烯,共軛炔低聚物等。同時,還是多種天然產物和具有生物活性化合物的骨架結構。含有1,3-二炔結構的一些化合物已被確定有著高效抗腫瘤、抗菌、抗炎和抗HIV活性。例如:從茵陳蒿中分離出來的傳統中藥成分19-(2-呋喃基)-5,7-十九碳二炔酸等。
文獻中報道過的合成不對稱共軛二炔的方法主要有以下幾種:
(一)2000年E.Negishi課題組利用Pd催化交叉偶聯選擇性合成非對稱1,3-共軛二炔。
該實驗方法雖然產率較高,但是卻需要分三步才能合成共軛二炔,反應較復雜。
(二)2008年Lei課題組報道了鈀催化的不對稱共軛二炔的合成。該反應以溴代苯乙炔和端基炔為底物,Pd(dba)2為催化劑,一種烯基膦為配體,三乙胺為有機弱堿,并加入CuI作助催化劑。
該反應底物的適用性較廣,有很好的選擇性,產率較高。但此方法使用了貴金屬催化劑和膦配體,合成成本高。
(三)2009年,Lei課題組在前期的工作基礎上,通過加入NiCl2·2H2O做催化劑并加入CuI作助催化劑,配以TMEDA絡合,使得能夠得到較高產率的交叉偶聯產物。
該實驗方法不僅使用雙金屬催化劑,并且加入TMEDA配體絡合,而且反應需要一種炔過量。
(四)2012年Ranu利用Cu-HAP配合物催化順式溴代苯乙烯合成非對稱的二炔。在此反應中,反式苯乙烯溴化物通常得到反式烯炔產物,而順式苯乙烯溴化物通過消除反應產生的炔和另一種炔烴的交叉偶聯,生成共軛二炔類化合物。
該實驗方法利用特定結構的銅配合物,此配合物的合成較復雜。
(五)2013年謝美華課題組報到了鎳催化的烷基格氏試劑與炔基砜的交叉偶聯反應合成不對稱1,3二炔。該合成中,1,4-二芳基1,3-二炔可以獲得中度至良好的產率,而對于烷基取代的1,3-二炔產率較低(30-54%)。
該實驗方法使用的烷基格氏試劑不穩定、不易操作。
(六)2014年Ranu課題組利用雙金屬無配體催化劑,通過Csp-Csp偶聯合成不對稱1,3-二炔。
該實驗方法底物適應性廣,但是使用了雙金屬催化劑。
綜上所述,現有技術合成共軛二炔的方法雖然較多,但這些方法有些需要經過較為復雜的合成步驟;有些用的反應物不穩定,操作有一定的難度;有些反應時間過長,而且反應產率較低;有些使用有機膦藥品易造成環境污染。更重要的是這類反應大都使用鈀、鎳等貴金屬或雙金屬為催化劑,反應成本較為昂貴而且不可回收,不適用于工業生產。
因此,提供一種不對稱共軛二炔的新型合成方法很有必要。
技術實現要素:
本發明的目的在于提供一種不對稱共軛二炔的合成方法,利用氬氣作保護氣,納米氧化銅催化順式烯溴和端基炔直接反應制備不對稱共軛二炔,合成方法成本低,原料易得,操作較為方便,效率高,使用范圍廣,適合多種底物反應,且催化劑能夠回收再利用。
本發明提供的一種不對稱共軛二炔的合成方法,包括以下步驟:
A、將催化劑與鄰菲羅啉和強堿混合后,在氬氣氛圍中加入順式烯溴、端基炔與溶劑,在140-150℃的條件下攪拌回流反應20-24h;
B、產物經萃取、干燥、濃縮后通過柱層析分離純化,得到不對稱共軛二炔。
步驟A中順式烯溴、端基炔、催化劑、強堿、鄰菲羅啉的摩爾比為1:1.2-1.5:0.05-0.1:2-2.5:0.15-0.2。
所述順式烯溴與機溶劑的用量比為:1:2-4mmol/mL。
進一步的,步驟A中相對于1mmol的順式烯溴,所述端基炔的用量1.2-1.5mmol,所述催化劑的用量為0.05-0.1mmol,所述強堿的用量為2-2.5mmol,所述鄰菲羅啉的用量為0.15-0.2mmol,所述有機溶劑的用量為2-4mL。
步驟A中所述的順式烯溴結構式為:
其中R1選自4-H、4-CH3、4-OCH3、3-OCH3、2-OCH3、4-Cl、3-Cl、2-Cl或4-CF3中的任意一個。
步驟A中所述端基炔的結構式為:
其中R2選自4-H、4-CH3、4-OCH3、3-OCH3、2-OCH3、4-Cl、3-Cl、2-Cl、4-Br、4-F、4-C(CH3)3中的任意一個。
步驟A中所述催化劑為梭形納米氧化銅、氧化銅納米粒子、片狀納米氧化銅、海參狀納米氧化銅或花狀納米氧化銅的任意一種,可以回收并循環使用,其中海參狀納米催化劑催化效果尤為顯著。
步驟A中各種催化劑的具體制備方法如下:
取40mL的蒸餾水加入到100mL燒杯中,稱取0.3194g的Cu(CH3COO)2·H2O固體置于燒杯中,在30℃磁力攪拌的情況下使其溶解,再緩慢滴入1M的NH3·H2O溶液調節pH值,將燒杯用保鮮膜包裹放于烘箱中,在65℃條件下反應24h冷卻至室溫,然后分別用蒸餾水和無水乙醇各清洗兩遍,分離離心,60℃條件下真空干燥5h即可制得不同形貌的納米氧化銅。此反應中pH為7得梭形納米氧化銅;pH為8得海參狀納米氧化銅pH為8.5得片狀納米氧化銅;pH為9.5得花狀納米氧化銅。
步驟A中所述氧化銅納米粒子的制備方法:取0.4525g的Cu(CH3COO)2溶于50mL無水乙醇中,待其溶解后置于容量60mL的聚四氟乙烯高壓釜中在150℃條件下反應20h,冷卻至室溫,再分別用蒸餾水清洗2遍和無水乙醇清洗3遍,分離離心,室溫條件下真空干燥得黑色粉末,即氧化銅納米粒子。
步驟A中所述強堿為氫氧化鈉、氫氧化鉀、叔丁醇鈉或碳酸銫中的任意一種。
步驟A中所述溶劑為二乙二醇二甲醚DGDE,N,N-二甲基甲酰胺DMF或二甲基亞砜DMSO中的任意一種。
步驟B具體為:將所得產物用石油醚、乙酸乙酯萃取,無水硫酸鈉干燥,減壓濃縮,得粗產物,以純石油醚為展開劑,通過柱層析分離得到不對稱1,3-共軛二炔。
所述不對稱共軛二炔的結構式為其中R1選自4-H、4-CH3、4-OCH3、3-OCH3、2-OCH3、4-Cl、3-Cl、2-Cl或4-CF3中的任意一個,R2選自4-H、4-CH3、4-OCH3、3-OCH3、2-OCH3、4-Cl、3-Cl、2-Cl、4-Br、4-F、4-C(CH3)3中的任意一個。
優選的,所述不對稱共軛二炔的合成方法,包括以下步驟:
A、將0.05mmol海參形納米氧化銅、0.15mmol鄰菲羅啉和2mmol氫氧化鈉混合后,在氬氣氛圍中加入1mmol順式烯溴、1.2mmol端基炔與3mL DGDE在145℃的條件下攪拌回流反應24h;
B、產物經萃取、干燥、濃縮后通過石油醚柱層析分離純化,得到不對稱共軛二炔。
與現有技術相比,本發明使用納米氧化銅催化劑,簡單易制,性能穩定,催化效果良好且能回收利用;原料以順式溴代烯烴和端基炔進行反應,反應條件溫和,不需要使用金屬格氏試劑,也沒有使用貴金屬或雙金屬催化劑;此反應體系成本低廉,簡單高效,環境友好,能夠合成高產率的不對稱共軛1,3二炔,有利于工業生產。
附圖說明
圖1為實施例1的核磁共振氫譜圖;
圖2為實施例1的核磁共振碳譜圖;
圖3為實施例2的核磁共振氫譜圖;
圖4為實施例2的核磁共振碳譜圖;
圖5為實施例3的核磁共振氫譜圖;
圖6為實施例3的核磁共振碳譜圖;
圖7為實施例4的核磁共振氫譜圖;
圖8為實施例4的核磁共振碳譜圖;
圖9為實施例5的核磁共振氫譜圖;
圖10為實施例5的核磁共振碳譜圖;
圖11為實施例6的核磁共振氫譜圖;
圖12為實施例6的核磁共振碳譜圖;
圖13為實施例7的核磁共振氫譜圖;
圖14為實施例7的核磁共振碳譜圖;
圖15為本發明的反應方程式。
具體實施方式
以下對本發明的具體實施方式進行詳細說明。應當理解的是,此處所描述的具體實施方式僅用于說明和解釋本發明,并不局限于這些實施例。實施例中,不同取代基的炔、鄰菲羅啉、氫氧化鈉、氫氧化鉀、叔丁醇鈉、碳酸銫、DGDE、DMF、DMSO為國藥集團化學試劑有限公司產品。
實施例1
一種不對稱共軛二炔的合成方法,包括以下步驟:
A、分別稱取0.05mmol的梭形納米氧化銅,0.15mmol鄰菲羅啉,2mmol氫氧化鈉放入潔凈干燥的反應管中,將反應管放置在無水無氧體系中,抽真空并通入氬氣循環三次,在氬氣保護下再依次加入1mmol順式-1-(2-溴乙烯基)-4-叔丁基苯,1.2mmol 1-乙炔基-4-甲氧基苯和3mL二乙二醇二甲醚,在140℃條件下攪拌回流20小時;
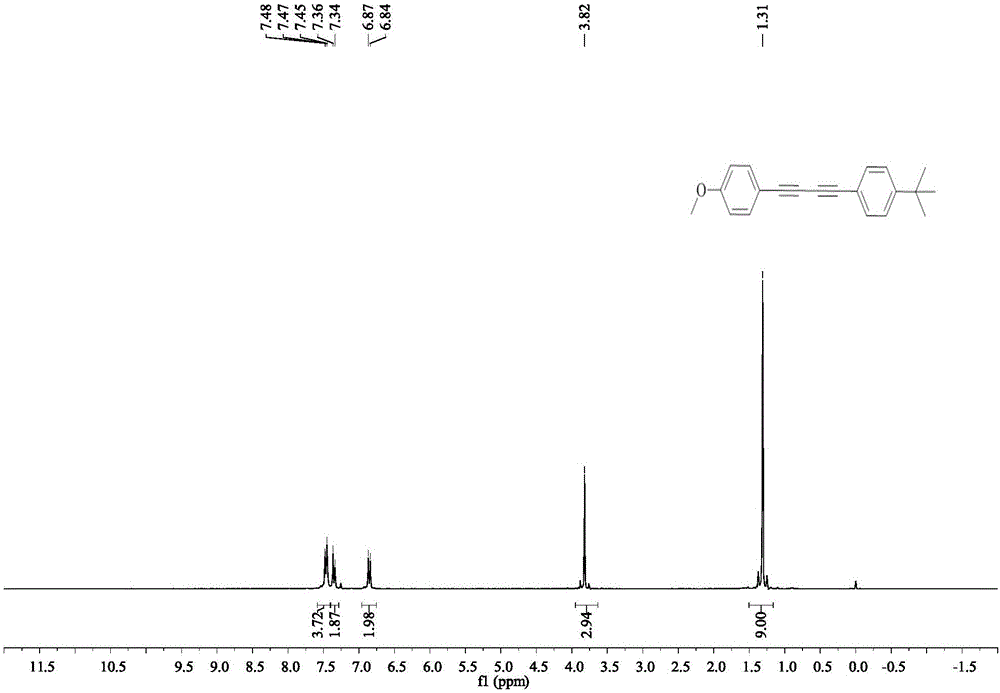
B、停止反應后冷卻至室溫,用石油醚萃取3次,每次10mL石油醚,并將所得有機相用無水硫酸鎂干燥,濃縮后,以石油醚作展開劑柱層析分離提純得白色固體即1-叔丁基-4-[4-(4-甲氧基苯基)丁烷-1,3-二炔基]苯,產率80%,熔點為88-90℃。
產品結構式:
產品的核磁共振氫譜數據:
1H NMR(300MHz,CDCl3):δ7.48-7.45(m,4H),7.35(d,J=8.2Hz,2H),6.86(d,J=8.6Hz,2H),3.82(s,3H),1.34(s,9H);
產品的核磁共振碳譜數據:
13C NMR(75MHz,CDCl3):δ160.3,152.5,134.1,132.2,125.5,118.9,114.1,113.8,81.4,79.1,73.5,72.9,55.4,34.9,31.1。
實施例2
一種不對稱共軛二炔的合成方法,包括以下步驟:
A、分別稱取0.05mmol的海參形納米氧化銅,0.15mmol鄰菲羅啉,2mmol氫氧化鉀放入潔凈干燥的反應管中,將反應管放置在無水無氧體系中,抽真空并通入氬氣循環三次,在氬氣保護下再依次加入1mmol順式-1-(2-溴乙烯基)-4-甲基苯,1.2mmol 1-乙炔基-4-氟基苯和3mL N,N-二甲基甲酰胺,在140℃條件下攪拌回流20小時。
B、停止反應后冷卻至室溫,用石油醚萃取3次,每次10mL石油醚,并將所得有機相用無水硫酸鎂干燥,濃縮。以石油醚作展開劑柱層析分離提純得白色固體即1-氟-4-[4-(4-甲基苯基)丁烷-1,3-二炔基]苯,產率85%,熔點為148-150℃。
產品結構式:
產品的核磁共振氫譜數據:
1H NMRδ7.53-7.49(m,2H),7.42(d,J=7.2Hz,2H),7.14(d,J=6.3Hz,2H),7.06-7.00(m,2H),2.36(s,3H);
產品的核磁共振碳譜數據:
13C NMR(CDCl3,75MHz):δ163.0(d,J=240.9Hz),139.6(d,J=15.3Hz),134.5,132.4,129.3,118.7(d,J=19.1Hz),115.9(d,J=21.8Hz),81.9,80.4,73.8,73.5,21.7.
實施例3
一種不對稱共軛二炔的合成方法,包括以下步驟:
A、分別稱取0.1mmol的片狀納米氧化銅,0.3mmol鄰菲羅啉,4mmol氫氧化鈉放入潔凈干燥的反應管中,將反應管放置在無水無氧體系中,抽真空并通入氬氣循環三次,在氬氣保護下再依次加入2mmol順式-1-(2-溴乙烯基)苯,2.4mmol 1-乙炔基-4-氟基苯和4mL二乙二醇二甲醚,在140℃條件下攪拌回流20小時;
B、停止反應后冷卻至室溫,用石油醚萃取3次,每次10mL石油醚,并將所得有機相用無水硫酸鎂干燥,濃縮。以石油醚作展開劑柱層析分離提純即白色固體即1-[4-(4-氟基苯基)丁烷-1,3-二炔基]苯,產率61%,熔點為121-123℃。
產品結構式:
產品的核磁共振氫譜數據:
1H NMR(CDCl3,300MHz):δ7.53(d,J=5.3Hz,4H),7.36(s,3H),7.04(t,J=8.4Hz,2H);
產品的核磁共振碳譜數據:
13C NMR(CDCl3,75MHz):δ163.0(d,J=250.5Hz),134.5(d,J=8.3Hz),132.5,129.2,128.4,121.6,117.9(d,J=3.0Hz),115.8(d,J=22.5Hz),81.5,80.4,73.7,73.7。
實施例4
一種不對稱共軛二炔的合成方法,包括以下步驟:
A、分別稱取0.5mmol的花狀納米氧化銅,1.5mmol鄰菲羅啉,20mmol氫氧化鈉放入潔凈干燥的反應管中,將反應管放置在無水無氧體系中,抽真空并通入氬氣循環三次。在氬氣保護下再依次加入10mmol順式-1-(2-溴乙烯基)-4-甲氧基苯,12mmol苯乙炔和20mL二乙二醇二甲醚,在150℃條件下攪拌回流20小時。
B、停止反應后冷卻至室溫,用石油醚萃取3次,每次40mL石油醚,并將所得有機相用無水硫酸鎂干燥,濃縮。以石油醚作展開劑柱層析分離提純得白色固體即1-[4-(4-甲氧基苯基)丁烷-1,3-二炔基]苯,產率61%,熔點為93-94℃。
產品結構式:
產品的核磁共振氫譜數據:
1H NMR(CDCl3,300MHz):δ7.46-7.53(m,4H),7.35(s,3H),6.86(d,J=8.2Hz,2H),3.83(s,3H);
產品的核磁共振碳譜數據:
13C NMR(CDCl3,75MHz):δ=160.3,134.0,132.3,129.0,128.4,121.9,114.1,113.6,81.8,81.0,74.2,72.7,55.2.
實施例5
一種不對稱共軛二炔的合成方法,包括以下步驟:
A、分別稱取0.5mmol的海參狀納米氧化銅,1.5mmol鄰菲羅啉,20mmol氫氧化鈉放入潔凈干燥的反應管中,將反應管放置在無水無氧體系中,抽真空并通入氬氣循環三次,在氬氣保護下再依次加入10mmol順式-1-(2-溴乙烯基)-4-甲基苯,12mmol 1-乙炔基-4-氟基苯和20mL二甲基亞砜,在140℃條件下攪拌回流20小時;
B、停止反應后冷卻至室溫,用石油醚萃取3次,每次10mL石油醚,并將所得有機相用無水硫酸鎂干燥,濃縮。以石油醚作展開劑柱層析分離提純得白色固體即1-氟-4-[4-(4-甲基苯基)丁烷-1,3-二炔基]苯,產率78%,熔點為148-150℃。
產品結構式:
產品的核磁共振氫譜數據:
1H NMRδ7.53-7.49(m,2H),7.42(d,J=7.2Hz,2H),7.14(d,J=6.3Hz,2H),7.06-7.00(m,2H),2.36(s,3H);
產品的核磁共振碳譜數據:
13C NMR(CDCl3,75MHz):δ163.0(d,J=240.9Hz),139.6(d,J=15.3Hz),134.5,132.4,129.3,118.7(d,J=19.1Hz),115.9(d,J=21.8Hz),81.9,80.4,73.8,73.5,21.7.
實施例6
一種不對稱共軛二炔的合成方法,包括以下步驟:
A、分別稱取0.025mmol的梭形納米氧化銅,0.075mmol鄰菲羅啉,1mmol叔丁醇鈉放入潔凈干燥的反應管中,將反應管放置在無水無氧體系中,抽真空并通入氬氣循環三次,在氬氣保護下再依次加入0.5mmol順式-1-(2-溴乙烯基)-4-氯基苯,0.6mmol 1-乙炔基-4甲氧基苯和2mL二乙二醇二甲醚,在140℃條件下攪拌回流24小時。
B、停止反應后冷卻至室溫,用石油醚萃取3次,每次10mL石油醚,并將所得有機相用無水硫酸鎂干燥,濃縮。以石油醚作展開劑柱層析分離提純得淡黃色固體即
1-氯-4–[4-(4-甲氧基苯基)丁烷-1,3-二炔基]苯,產率80%,熔點為160-162℃。
產品結構式:
產品的核磁共振氫譜數據:
1H NMR(300MHz,CDCl3):δ7.49-7.43(m,4H),7.30(d,J=9.0Hz,2H),6.87(d,J=9.0Hz,2H),3.83(s,3H).
產品的核磁共振氫譜數據:
13C NMR(75MHz,CDCl3):δ160.1,135.1,134.2,133.6,128.8,120.5,114.2,113.5,82.4,79.8,75.2,72.5,55.4.
實施例7
一種不對稱共軛二炔的合成方法,包括以下步驟:
A、分別稱取0.1mmol的氧化銅納米粒子,0.15mmol鄰菲羅啉,2mmol碳酸銫放入潔凈干燥的反應管中,將反應管放置在無水無氧體系中,抽真空并通入氬氣循環三次,在氬氣保護下再依次加入1mmol順式-1-(2-溴乙烯基)-4氯基苯,1.2mmol苯乙炔和3mL二乙二醇二甲醚,在150℃條件下攪拌回流20小時;
B、停止反應后冷卻至室溫,用石油醚萃取3次,每次10mL石油醚,并將所得有機相用無水硫酸鎂干燥,濃縮。以石油醚作展開劑柱層析分離提純得白色固體即1-[4-(4-氯基苯基)丁烷-1,3-二炔基]苯,產率80%,熔點為151-152℃。
產品結構式:
產品的核磁共振氫譜數據:
1H NMR(300MHz,CDCl3):δ7.54-7.30(m,9H).
產品的核磁共振碳譜數據:
13C NMR(75MHz,CDCl3):δ135.4,133.7,132.5,128.9,128.5,121.6,120.3,82.1,80.3,74.9,73.6.
實施例8
一種不對稱共軛二炔的合成方法,包括以下步驟:
A、分別稱取0.05mmol的花狀納米氧化銅,0.15mmol鄰菲羅啉,2mmol氫氧化鉀放入潔凈干燥的反應管中,將反應管放置在無水無氧體系中,抽真空并通入氬氣循環三次,在氬氣保護下再依次加入1mmol順式-1-(2-溴乙烯基)-4-甲氧基苯,1.2mmol 1-乙炔基-4-叔丁基苯和3mL二乙二醇二甲醚,在140℃條件下攪拌回流20小時;
B、停止反應后冷卻至室溫,用石油醚萃取3次,每次10mL石油醚,并將所得有機相用無水硫酸鎂干燥,濃縮,以石油醚作展開劑柱層析分離提純得白色固體即1-叔丁基-4-[4-(4-甲氧基苯基)丁烷-1,3-二炔基]苯,產率80%,熔點為88-90℃。
產品結構式:
1H NMR(300MHz,CDCl3):δ7.48-7.45(m,4H),7.35(d,J=8.2Hz,2H),6.86(d,J=8.6Hz,2H),3.82(s,3H),1.34(s,9H);
13C NMR(75MHz,CDCl3):δ160.3,152.5,134.1,132.2,125.5,118.9,114.1,113.8,81.4,79.1,73.5,72.9,55.4,34.9,31.1。
- 還沒有人留言評論。精彩留言會獲得點贊!