胺碘酮雜質G的合成方法及胺碘酮雜質G的應用與流程
本發明涉及化學合成
技術領域:
,尤其是涉及一種胺碘酮雜質G的合成方法及胺碘酮雜質G的應用。
背景技術:
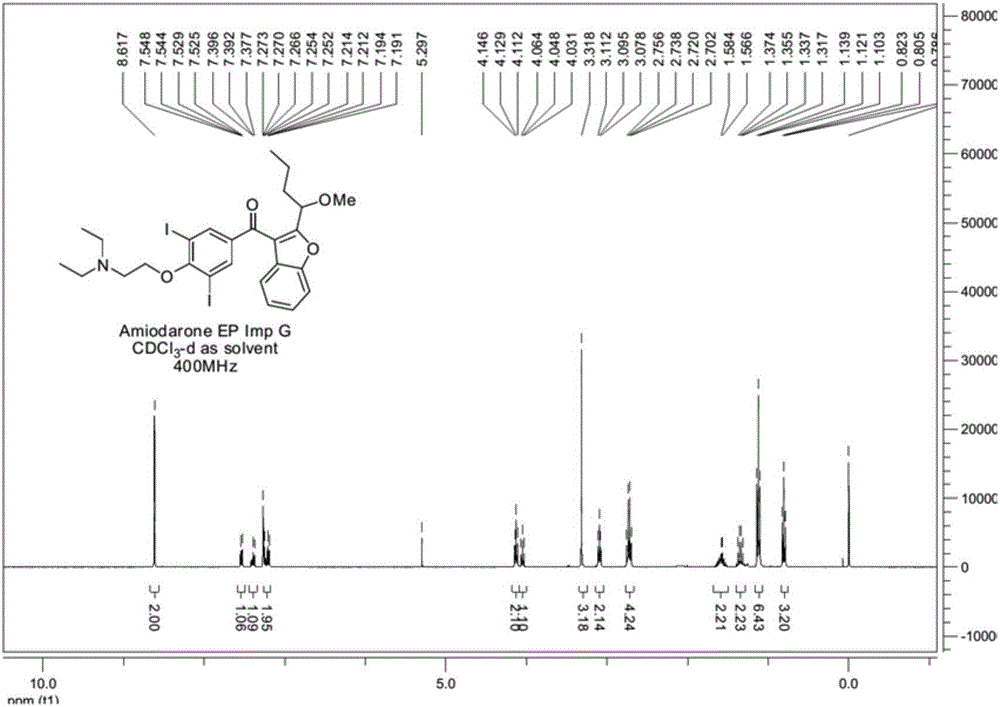
:胺碘酮,又稱安碘達隆,其英文名稱為Amiodarone。胺碘酮屬于III類抗心律失常藥,口服適用于房性早搏及室性早搏;對反復性陣發性室上性心動過速、心房顫動、心房撲動、室性心動過速及室顫可防止反復發作,也可防止預激綜合征伴室上性心律失常的發作及心房顫動或心房撲動電轉復后的維持治療。胺碘酮的質量標準在多國藥典中均中有所記載,英國藥典更是明確指出了胺碘酮雜質G的具體結構。現有技術中,已經公開了多種胺碘酮質量控制的檢測方法,以檢測包括胺碘酮雜質G在內的多種雜質的含量。但是現有技術中,尚未公開胺碘酮雜質G的高效合成方法。胺碘酮雜質G是胺碘酮質量控制中非常重要的雜質,對胺碘酮雜質的相關研究意義重大。因此,提供一種胺碘酮雜質G的高效合成方法,能夠彌補現有技術中的不足,為胺碘酮的質量控制提供合格、廉潔、易得的對照品,為安全用藥提供重要的指導意義。因此,特提出本發明。技術實現要素:本發明的目的在于提供一種胺碘酮雜質G的合成方法,以緩解現有技術中的不足,從而為胺碘酮雜質G的質量控制提供雜質對照品。本發明提供了一種胺碘酮雜質G的合成方法,所述合成方法包括以下步驟:(1)在惰性氣體保護下,式I所示結構的化合物和正丁基鋰,在反應介質中進行第一反應;(2)待所述第一反應結束后,向所述第一反應的反應液中,加入式II所示結構的化合物,進行第二反應,得到式III所示結構的化合物,即胺碘酮雜質G,所述胺碘酮雜質為式III所示結構的化合物。進一步的,步驟(2)中,所述第二反應中還加入催化劑,所述催化劑為無水三氯化鐵。進一步的,步驟(1)中,所述反應介質為四氫呋喃。進一步的,步驟(1)中,所述第一反應的反應溫度為-70℃以下。進一步的,步驟(1)中,所述惰性氣體為氮氣或者氬氣。進一步的,步驟(2)中,所述第二反應的反應溫度為-70~25℃。進一步的,所述式I所示結構的化合物和所述式II所示結構的化合物的質量比為1.8~2.2:0.8~1.2,優選2:1。進一步的,步驟(1)中,所述第一反應的反應時間為10~30min。進一步的,步驟(2)中,所述第二反應的反應時間為2~6h。另外本發明的另一目的在于提供一種按照所述的合成方法合成的所述胺碘酮雜質G在制備胺碘酮雜質對照品中的應用,以為胺碘酮的質量控制提供雜質對照品。本發明提供的胺碘酮雜質G的合成方法以式I和式II所示結構的化合物為原料,制備得到純度高、結構正確的胺碘酮雜質G。該合成方法成本低、產率高、操作簡單。另外,本發明提供了按照所述方法合成的胺碘酮雜質G制備胺碘酮雜質對照品中的應用,為胺碘酮的質量控制提供了合格、廉價、易得的雜質對照品。附圖說明為了更清楚地說明本發明具體實施方式或現有技術中的技術方案,下面將對具體實施方式或現有技術描述中所需要使用的附圖作簡單地介紹,顯而易見地,下面描述中的附圖是本發明的一些實施方式,對于本領域普通技術人員來講,在不付出創造性勞動的前提下,還可以根據這些附圖獲得其他的附圖。圖1為本發明實施例1合成的產物的氫譜圖。具體實施方式下面將結合附圖對本發明的技術方案進行清楚、完整地描述,顯然,所描述的實施例是本發明一部分實施例,而不是全部的實施例。基于本發明中的實施例,本領域普通技術人員在沒有做出創造性勞動前提下所獲得的所有其他實施例,都屬于本發明保護的范圍。本發明提供了一種胺碘酮雜質G的合成方法,包括以下步驟:(1)在惰性氣體保護下,式I所示結構的化合物和正丁基鋰,在反應介質中進行第一反應;(2)待第一反應結束后,向第一反應的反應液中,加入式II所示結構的化合物,進行第二反應,得到式III所示結構的化合物,即胺碘酮雜質G。其中,式I所示結構的化合物,化學名稱為:3-溴-2-(1-甲氧基丁基)苯并呋喃;式II所示結構的化合物,化學名稱為:4-(2-(二乙氨基)乙氧基)-3,5-二碘苯甲酰氯。需要注意的是,本發明提供的合成方法需要在無水條件下進行。在一個優選的實施方式中,步驟(2)還包括對反應生成的式III所示結構的化合物的純化,具體為:第二反應結束后,將反應液過濾,濾液減壓濃縮,將濃縮物用HPLC純化,純化后的物質繼續濃縮干燥得到的物質即為式III所示結構的化合物,即胺碘酮雜質G。在另一個優選的實施方式中,式I所示結構的化合物和式II所示結構的化合物的質量比為1.8~2.2:0.8~1.2,優選2:1。在另一個優選的實施方式中,式I所示結構的化合物、四氫呋喃、式II所示結構的化合物、無水三氯化鐵的質量體積比為27~33:300~600:12~18:0.5~1.5(g:mL:g:g),例如可以為27:300:12:0.5(g:mL:g:g)、33:600:18:1.5(g:mL:g:g)、28:400:14:0.8(g:mL:g:g)、32:500:16:1.2(g:mL:g:g)或者30:450:15:1(g:mL:g:g),其中優選為30:450:15:1(g:mL:g:g)。在另一個優選的實施方式中,正丁基鋰為正丁基鋰的四氫呋喃溶液。另外,本發明中涉及的化合物的中英文對照如表1所示:表1化合物縮寫對照表英文縮寫中文名稱THF四氫呋喃MTBE甲基叔丁基醚n-BuLi正丁基鋰PE石油醚EA乙酸乙酯NaH氫化鈉DCM二氯甲烷ACN乙腈HAc醋酸H5IO6高碘酸Cs2CO3碳酸銫DMFN,N-二甲基甲酰胺SOCl2氯化亞砜原料合成本發明所用的原料:式I所示結構的化合物及式II所示結構的化合物,均由發明人自主合成,合成方法如下。1.式I所示結構的化合物的合成路線步驟一:將30g2,3-苯并呋喃加入到100ml三口瓶中,加入300mlTHF,冷卻到-65℃,滴加100ml2.5M的正丁基鋰,控溫小于-60℃,加完控溫小于65℃反應1h。然后滴加9g正丁醛的THF溶液,控溫小于-60℃,加完反應半小時。TLC點板顯示反應完全,其中TLC展開劑為PE:EA=3:1。將反應液倒入到300ml水中,用MTBE萃取(500mlMTBE萃取兩次),有機相干燥旋干,得到40g棕色油,即為式M1-1所示結構的化合物,收率為83%。步驟二:將40g式M1-1所示結構的化合物,加入到1L三口瓶中,加入500mlDMF,冷卻到10℃,分批次加入10g氫化鈉,控溫小于20℃,加完后,在20℃反應20min,滴加44g碘甲烷,控溫小于20℃,加完20℃反應0.5h。TLC點板顯示反應完全,其中TLC展開劑為PE:EA=10:1。將反應液倒入到2L水中,用MTBE萃取(500mlMTBE萃取兩次),有機相干燥選干,得到40g棕色油,即為式M1-2所示結構的化合物,收率為93%。步驟三:將40g式M1-2所示結構的化合物,加入到1L三口瓶中,加入200mlDCM和200ml乙腈,加入40gN-溴代琥珀酰亞胺,室溫反應過夜。TLC點板顯示反應完全,其中TLC展開劑為PE:EA=10:1。反應液加硅膠旋干過柱(PE),得到40g淡黃色固體,即為式I所示結構的化合物,收率為72%。2.式II所示結構的化合物的合成路線步驟一:將30.4g對羥基苯甲酸甲酯,加入到2L單口瓶中,加入1L醋酸、500ml水、42.8g高碘酸和66.4g碘化鉀,60℃反應過夜。TLC點板顯示反應完全,其中TLC展開劑為PE:EA=5:1。將反應液冷卻到室溫,加入到2L水中,過濾,固體用紅外燈干燥,得到80g黃色固體,即為式M2-1所示結構的化合物,收率為99%。步驟二:將20.8g式M2-1所示結構的化合物,加入到1L三口瓶中,加入500mlDMF和40.6g碳酸銫,室溫反應10min,然后分批次加入11.1gN,N-二乙基氯乙胺鹽酸鹽,加完后室溫反應過夜。TLC點板顯示反應完全,其中TLC展開劑為PE:EA=0:1。將反應液倒入到2L水中,加入500mlEA,攪拌10min過濾,濾液分液,有機相用醋酸水溶液洗一遍,所得水相再用碳酸鉀調PH=8,然后用EA萃取(200mlEA萃取兩次),有機相選干,得到5g淺棕色油,即為式M2-2所示結構的化合物,收率為20%。步驟三:將5gM4加入到250ml單口瓶中,加入30ml濃鹽酸,100℃反應過夜,將反應液冷卻到室溫,加入20mlEA,攪拌10min,過濾,固體用紅外燈干燥。將所得酸加入到250ml單口瓶中,加入50ml二氯乙烷,然后加入8g氯化亞砜和0.5mlDMF,升溫到80℃回流反應三小時。反應液直接選干,加入20mlEA打漿,過濾,固體用旋轉蒸發儀蒸干,得到4g淺棕色固體,即為式II所示結構的化合物,收率為80%。為了有助于更清楚的理解本發明的內容,現結合具體實施例及對比例詳細介紹如下:如未明確指出,本發明所用的試劑為常用試劑,購自于市場,所涉及的室溫為25℃。實施例1向250mL三口瓶中加入90mL四氫呋喃和6g式I所示結構的化合物加入,氮氣保護,冷卻到-70℃,滴加10mL2.5mol/L的正丁基鋰溶液,滴加時需保證控制溫度小于-65℃,加完后在-70℃反應10min。然后加入3g式II所示結構的化合物和0.2g無水氯化鐵,然后回到室溫反應2h。對反應結束后的反應液進行純化,用旋轉蒸發儀減壓濃縮干,設置壓強為0.09Mpa,濃縮溫度為35℃,濃縮后的溶液用HPLC分離純化,HPLC的具體參數設置如下:色譜柱:DAC100-10C18流動相:A相,0.1%甲酸;B相,甲醇流速:150mL/min檢測波長:240nm梯度設置:時間(min)0105080B相(%)204090100產物的出峰時間在50min左右,取產物峰對應的溶液用旋轉蒸發儀減壓濃縮,設置壓強為0.09Mpa,濃縮溫度為40℃,最終得到3.1g淺棕色油狀物,即為胺碘酮雜質G,產率為77.7%。圖1所示為實施例1得到的胺碘酮雜質G的氫譜圖,確定了目標產物。實施例2向250mL三口瓶中加入67mL四氫呋喃和6g式I所示結構的化合物加入,氮氣保護,冷卻到-70℃,滴加10mL2.5mol/L的正丁基鋰溶液,滴加時需保證控制溫度小于-65℃,加完后在-70℃反應10min。然后加入2.7g式II所示結構的化合物和0.1g無水氯化鐵,然后回到室溫反應2h。按照實施例1中提供的純化方法,對反應液進行HPLC純化、減壓濃縮,最終得到2.7g淺棕色油狀物,即為胺碘酮雜質G,產率為75%。實施例3向250mL三口瓶中加入109mL四氫呋喃和6g式I所示結構的化合物加入,氮氣保護,冷卻到-70℃,滴加10mL2.5mol/L的正丁基鋰溶液,滴加時需保證控制溫度小于-65℃,加完后在-70℃反應10min。然后加入3.3g式II所示結構的化合物和0.27g無水氯化鐵,然后回到室溫反應2h。按照實施例1中提供的純化方法,對反應液進行HPLC純化、減壓濃縮,最終得到3.3g淺棕色油狀物,即為胺碘酮雜質G,產率為75.2%。對比例1向250mL三口瓶中加入40mL四氫呋喃和6g式I所示結構的化合物加入,氮氣保護,冷卻到-70℃,滴加10mL2.5mol/L的正丁基鋰溶液,滴加時需保證控制溫度小于-65℃,加完后在-70℃反應10min。然后加入1g式II所示結構的化合物和0.2g無水氯化鐵,然后回到室溫反應2h。按照實施例1中提供的純化方法,對反應液進行HPLC純化、減壓濃縮,最終得到0.4g淺棕色油狀物,即為胺碘酮雜質G,產率為30%。對比例2向500mL三口瓶中加入180mL四氫呋喃和3g式I所示結構的化合物加入,氮氣保護,冷卻到-70℃,滴加5mL2.5mol/L的正丁基鋰溶液,滴加時需保證控制溫度小于-65℃,加完后在-70℃反應10min。然后加入6g式II所示結構的化合物和0.03g無水氯化鐵,然后回到室溫反應2h。按照實施例1中提供的純化方法,對反應液進行HPLC純化、減壓濃縮,最終得到4.6g淺棕色油狀物,即為胺碘酮雜質G,產率為32.2%。為了更清楚的比較,將實施例1-3和對比例1-2的變量和實驗結果列于表2:表2實施例1-3及對比例1-2的變量及產率式I所示結構的化合物:四氫呋喃:式II所示結構的化合物:無水三氯化鐵產率實施例130:450:15:1(g:mL:g:g)77.7%實施例227:300:12:0.5(g:mL:g:g)75%實施例333:600:18:1.5(g:mL:g:g)75.2%對比例130:200:5:1(g:mL:g:g)30%對比例210:600:20:0.1(g:mL:g:g)32.2%實施例4向250mL三口瓶中加入90mL四氫呋喃和6g式I所示結構的化合物加入,氮氣保護,冷卻到-70℃,滴加10mL2.5mol/L的正丁基鋰溶液,滴加時需保證控制溫度小于-65℃,加完后在-75℃反應10min。然后加入3g式II所示結構的化合物和0.2g無水氯化鐵,然后回到室溫反應2h。按照實施例1中提供的純化方法,對反應液進行HPLC純化、減壓濃縮,最終得到3.1g淺棕色油狀物,即為胺碘酮雜質G,產率為80.2%。對比例3向250mL三口瓶中加入90mL四氫呋喃和6g式I所示結構的化合物加入,氮氣保護,冷卻到-70℃,滴加10mL2.5mol/L的正丁基鋰溶液,滴加時需保證控制溫度小于-65℃,加完后在-60℃反應10min。然后加入3g式II所示結構的化合物和0.2g無水氯化鐵,然后回到室溫反應2h。按照實施例1中提供的純化方法,對反應液進行HPLC純化、減壓濃縮,最終得到3.1g淺棕色油狀物,即為胺碘酮雜質G,產率為10.0%。為了更清楚的比較,將實施例1、4和對比例3的變量和實驗結果列于表3:表3實施例1、4及對比例3的變量及產率第一反應溫度產率實施例1-70℃77.7%實施例4-75℃80.2%對比例3-60℃10.0%對比例4向250mL三口瓶中加入90mL乙醚和6g式I所示結構的化合物加入,氮氣保護,冷卻到-70℃,滴加10mL2.5mol/L的正丁基鋰溶液,滴加時需保證控制溫度小于-65℃,加完后在-60℃反應10min。然后加入3g式II所示結構的化合物和0.2g無水氯化鐵,然后回到室溫反應2h。按照實施例1中提供的純化方法,對反應液進行HPLC純化、減壓濃縮,最終得到0.2g淺棕色油狀物,即為胺碘酮雜質G,產率為5.0%。對比例5向250mL三口瓶中加入90mL環己烷和6g式I所示結構的化合物加入,氮氣保護,冷卻到-70℃,滴加10mL2.5mol/L的正丁基鋰溶液,滴加時需保證控制溫度小于-65℃,加完后在-60℃反應10min。然后加入3g式II所示結構的化合物和0.2g無水氯化鐵,然后回到室溫反應2h。按照實施例1中提供的純化方法,對反應液進行HPLC純化、減壓濃縮,最終得到0.3g淺棕色油狀物,即為胺碘酮雜質G,產率為7.5%。為了更清楚的比較,將實施例1和對比例4、5的變量和實驗結果列于表4:表4實施例1及對比例4、5的變量及產率溶劑產率實施例1四氫呋喃77.7%對比例4乙醚5.0%對比例5環己烷7.5%對比例6向250mL三口瓶中加入90mL四氫呋喃和6g式I所示結構的化合物加入,氮氣保護,-70℃反應10min。然后加入3g式II所示結構的化合物和0.2g無水氯化鐵,然后回到室溫反應2h。按照實施例1的HPLC方法對反應液進行處理,HPLC結果顯示,并沒有明顯的產物峰出現,表明按照對比例1提供的方法,不能合成目標產物。為了更清楚的比較,將實施例1和對比例6的變量和實驗結果列于表5:表5實施例1及對比例6的變量及產率試劑產率實施例1正丁基鋰77.7%對比例6—0%以上實驗表明,本發明提供的胺碘酮雜質G的合成方法,在正丁基鋰的作用下,以式I所示結構的化合物及式II所示結構的化合物為原料,選用合適的反應介質、反應條件及反應劑量,高效的合成了結構正確的胺碘酮雜質G。本發明提供的合成方法操作簡單、產率高,具有極大的商業價值和市場前景。另外,按照本發明提供的合成方法獲得的胺碘酮雜質G能夠用于制備胺碘酮雜質對照品,為胺碘酮的質量控制提供了廉價、易得的對照品。最后應說明的是:以上各實施例僅用以說明本發明的技術方案,而非對其限制;盡管參照前述各實施例對本發明進行了詳細的說明,本領域的普通技術人員應當理解:其依然可以對前述各實施例所記載的技術方案進行修改,或者對其中部分或者全部技術特征進行等同替換;而這些修改或者替換,并不使相應技術方案的本質脫離本發明各實施例技術方案的范圍。當前第1頁1 2 3
相關技術
網友詢問留言
已有0條留言
- 還沒有人留言評論。精彩留言會獲得點贊!
1