一種修復酵母染色體結構異常的方法與流程
本發明涉及基因組工程領域,具體涉及一種修復酵母染色體結構異常的方法。
背景技術:
染色體結構異常是指染色體非正常的發生斷裂、重組或互換后產生染色體畸變。染色體結構異常的類型可以分為DNA拷貝數變異、大片段插入、大片段復制、大片段翻轉、染色體移位等。染色體結構異常廣泛發生在絕大多數物種中,很多染色體結構異常與細胞衰老、染色體遺傳性疾病、癌癥等疾病密切相關。例如,貓叫綜合癥是人的第五號染色體部分缺失引起的遺傳病,果蠅的缺刻翅也是由于一段染色體的缺失造成的;果蠅的棒眼現象是X染色體上的部分片段重復引起的;第九號染色體長臂的倒置會造成女性習慣性流產。染色體結構變異可能來自于遺傳性的變異也可能是由于射線、化學試劑、溫度等外部環境的誘導或者生物體內代謝失衡、細胞衰老凋亡等引起的病變性質的變異,還可能來自于分子生物學的操作比如基因組編輯和大尺度DNA的合成等。大多數的染色體結構變異對生物體是不利的,會引起細胞生長缺陷,嚴重的甚至會致死。
酵母是與人類活動關系最密切的微生物之一,比較基因組學的研究表明不同種屬的釀酒酵母之間有大量的染色體結構變異,這些染色體的結構差異一方面導致了不同物種的區分,另一方面也使不同類型的酵母擁有不同的生長特性。比如有耐高溫耐抑制劑的釀酒酵母,也有高產乙醇的工業酵母菌株。和其他物種相似,酵母的染色體結構變異絕大多數也是不利的,需要對其進行修復。現有的染色體修復技術涉及修復的DNA尺度都比較小,比如利用CRISR-Cas9技術可以較好的實現點突變的修復或者和同源重組技術相結合可以實現數Kb長度的DNA序列的修復,但外源長DNA片段的合成和較低的轉化效率使得CRISPR-Cas9技術一般局限于數Kb長度DNA序列的改變,對于數百Kb長度的DNA無法有效修復。目前針對數百Kb長度的DNA修復技術鮮有報道。因此,需要開發一種高效準確的修復酵母染色體結構異常的技術。
技術實現要素:
有鑒于此,本發明的目的在于提供一種修復酵母染色體結構異常的方法,使得所述方法能夠更加簡便和快速的修復酵母異常染色體,并擴大DNA的修復尺度至數百Kb。
為了實現上述目的,本發明提供如下技術方案:
一種修復酵母染色體結構異常的方法,包括:
步驟1、確認變異酵母的染色體結構異常區域;
步驟2、變異酵母染色體上,在與著絲粒最接近的染色體結構異常區域的近著絲粒端朝著絲粒方向延伸的區域插入篩選標簽基因1;正常酵母染色體上,在與篩選標簽基因1相對應的位置處朝著絲粒方向延伸的區域插入篩選標記標簽2;所述篩選標簽基因1與篩選標簽基因2不同,所述正常酵母在其與變異酵母的染色體結構異常區域相對應區域上具有正常染色體;
步驟3、將不同交配型的變異酵母和正常酵母交配形成二倍體酵母,然后進行減數分裂獲得四分體,對四分體進行拆分獲得孢子;
步驟4、根據篩選標簽基因1和篩選標簽基因2分別設置對應的篩選標簽培養基,將所述孢子分別接種到兩種篩選標記培養基上,兩種篩選培養基上均無法正常生長的孢子為已經修復的酵母。
由于借助CRISR-Cas9技術對異常染色體修復時存在DNA修復尺度較小,且比較繁瑣的缺陷,本發明結合酵母高效的同源重組機制,通過篩選標記和減數分裂實現大尺度酵母染色體的高效修復。
為了方便理解本發明的技術思路,在圖1所示的染色體修復示意圖的基礎上對本發明方法的原理進行闡述(在圖1中,篩選標簽1和2分別選擇URA、LEU,簡寫為U和L,陰影部分表示染色體結構異常區域,交叉區域為同源重組鏈交換區域,即U和L之間的區域):
本發明利用酵母細胞減數分裂時非姐妹染色單體的同源重組實現結構異常的染色體和結構正常的染色體之間的鏈交換。在單倍體酵母菌株中出現染色體結構變異的末端位置插入URA基因。在染色體結構正常的單倍體酵母中(與染色體結構變異的單倍體酵母不同的交配型,具有完全正常的染色體),將LEU基因插入到與染色體結構異常區域對應的鏈交換區域。將上述兩個不同交配型的菌株交配形成二倍體酵母,在生孢培養基中培養二倍體菌株使其生孢。使用酵母顯微操作儀對生孢的四分體細胞進行拆分,之后將生長的單倍體孢子接種到SC-URA(缺URA培養基)和SC-LEU(卻LEU培養基)的平板來篩選在鏈交換區發生鏈交換的孢子,只有在設定區間發生了鏈交換才會引起兩個標簽基因位置的變換,根據結果可知,形成的四分體經過拆分后會出現四種單倍體孢子形式,其中一種就是不含任何篩選標簽的已經完全修復的菌株,既不能在SC-URA平板上生長也不能在SC-LEU平板上生長的單倍體孢子就是所需要的菌株。
對于變異酵母的異常染色體區域的定位方法可按照本領域常規的方法進行,在本發明具體實施中,本發明步驟1為通過脈沖場凝膠電泳和全基因組測序分析確認變異酵母的染色體結構異常區域。
本發明中篩選標簽基因1的插入位置是一個區間范圍,從與著絲粒最接近的染色體結構異常區域的近著絲粒端開始至著絲粒這一段區域都可以作為篩選標簽基因1的插入位置,以圖1為例說明,這一區域就是篩選標簽U到著絲粒之間的區域,考慮到后續篩選標簽基因2的插入,一般不選擇極為靠近著絲粒的區域。在本發明具體實施過程中,篩選標簽基因1的插入位置通常是距染色體結構異常區域的近著絲粒端100-1000bp長度位置處,即所述與著絲粒最接近的染色體結構異常區域的近著絲粒端朝著絲粒方向延伸的區域為與著絲粒最接近的染色體結構異常區域近著絲粒端朝著絲粒方向延伸100-1000bp的區域。
在本發明方法中,選擇的正常酵母具備完全正常的染色體,與變異酵母除了在交配型和染色體結構異常區域的差別外(交配型不同是為了能夠形成二倍體),其他位置的染色體基本相同。如此,變異酵母染色體上插入篩選標簽基因1的位置,在正常酵母染色體上也會存在相對應的位置(為表述方便,該位置稱為篩選標簽基因1對應位置),從篩選標簽基因1對應位置開始至著絲粒這一個區域為篩選標簽基因2的插入位置,以圖1為例說明,這一區域就是篩選標簽U對應到正常酵母染色體上的位置到著絲粒之間的區域,考慮到后續快捷拆分到正確四分體,篩選標簽基因2的插入位置通常是距篩選標簽基因1對應位置越遠越好,這樣預期在鏈交換區發生鏈交換的幾率則呈正向相關,拆分到正確的四分體也就更加快捷,由于二倍體酵母經過培養,其形成的眾多四分體中存在不計其數的各種鏈交換情形,本發明所預想的鏈交換情形通常均會發生。
在本發明具體實施過程中,篩選標簽基因2的插入位置通常是距篩選標簽基因1對應位置10-100kb的區域長度位置處。即所述在與篩選標簽基因1相對應的位置處朝著絲粒方向延伸的區域為在與篩選標簽基因1相對應的位置處朝著絲粒方向延伸10-100kb的區域。
對于篩選標簽基因的插入,可通過組裝上游500bp同源臂、篩選標簽基因、下游500bp同源臂三個片段構成待整合的外源片段,通過酵母同源重組整合到預期位置。
本發明中所提及的篩選標簽基因1和篩選標簽基因2可選自微生物領域中經常用于篩選目標微生物所采用的篩選標簽基因,如營養缺陷標簽基因和抗藥標簽基因。所述營養缺陷標簽一般為氨基酸缺陷標簽例如URA、LEU和HIS;所述抗藥標簽一般選自KanMX、NAT和Hyg。
在根據篩選標簽基因1和篩選標簽基因2分別設置對應的篩選標簽培養基時,本領域技術人員可根據選擇的標簽輕易設置,如采用URA篩選標簽,則對應的篩選標簽培養基為缺URA培養基(SC-URA),采用KanMX篩選標簽則對應的篩選標簽培養基為含有G418的培養基。
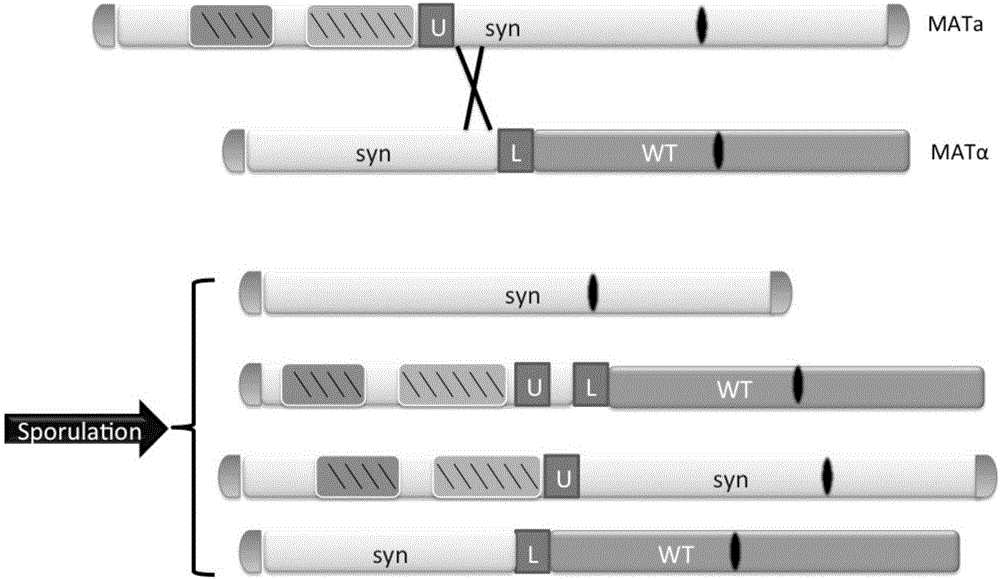
在酵母功能基因組學領域,隨著酵母基因組信息的被揭示,合成型染色體的酵母(包含全合成型染色體酵母和半合成型染色體酵母)因其方便性被廣泛使用,然而其同樣會產生染色體變異,需要進行修復。本發明所述方法同樣適用于合成型染色體酵母的染色體結構異常修復,此時本發明所述變異酵母為全合成型染色體或半合成型染色體的酵母,所述正常酵母為半合成型染色體酵母或全合成型染色體酵母;所述正常酵母在變異酵母染色體結構異常區域相對應區域上具有正常的合成型染色體;所述合成型染色體與野生型染色體在每個功能基因上序列存在微小差別(用于識別合成型染色體),高度相同,同時能夠表達相同蛋白。具體的染色體修復示意圖見圖2。
在本發明具體實施方式中,本發明以出現染色體結構異常的全合成型染色體釀酒酵母R-2為對象進行修復,脈沖場凝膠電泳對釀酒酵母R-2菌進行核型分析,可以看到菌株R-2和野生型對照菌株BY4741相比,十號染色體對應的條帶明顯向上發生了遷移。在全基因組測序分析菌株R-2后,通過測序深度圖發現有兩處區域有大片段復制重復。一處主要發生在megachunk C區域,涉及序列長度為101kb,為連續發生的三拷貝的復制。第二處主要發生在megachunk E區域,涉及序列長度為147kb,為二拷貝和四拷貝交錯排布的復制。綜合計算R-2菌株十號染色體的長度比野生型長度長171kb。
然后在單倍體酵母菌株R-2中出現染色體結構變異的末端位置插入URA基因。在染色體結構正常的單倍體半合成型染色體酵母BY4742中(與菌株R-2不同的交配型),將LEU基因插入到與染色體結構異常區域對應的鏈交換區域。將上述兩個不同交配型的菌株R-2和BY4742交配形成二倍體酵母,在生孢培養基中培養二倍體菌株使其生孢。使用酵母顯微操作儀對生孢的四分體細胞進行拆分,拆分的單倍體細胞按照四分體的來源整齊排布在YPD平板上培養2天,之后將生長的單倍體孢子平板翻印到SC-URA和SC-LEU的平板來篩選在設定區間發生鏈交換的孢子。挑取既不能在SC-URA平板上生長也不能在SC-LEU平板上生長的單倍體孢子,只有在設定區間發生了鏈交換才會引起兩個標記基因位置的變換。挑取通過表型驗證的單倍體孢子,利用PCR或其他檢測方法驗證染色體結構異常是否被修復。
由以上技術方案可知,本發明利用酵母高效的同源重組機制以及篩選標簽,快速篩選在設計區域發生鏈交換的酵母染色體,實現染色體結構異常的序列與正常染色體序列的交換,從而達到大尺度高效修復酵母染色體結構變異的目的。
附圖說明
圖1所示為本發明修復染色體結構異常的示意圖;其中,MATa與MATα代表酵母中的兩種交配型,陰影部分為染色體結構異常區域,U和L分別代表URA基因和LEU基因,交叉位置為鏈交換區;
圖2所示為本發明修復染色體結構異常的示意圖;其中,MATa與MATα代表酵母中的兩種交配型,陰影部分為染色體結構異常區域,U和L分別代表URA基因和LEU基因,syn表示合成型染色體,WT表示野生型染色體,sporulation表示生孢,交叉位置為鏈交換區;
圖3所示為脈沖場凝膠電泳;其中,羅馬數字表示染色體編號,BY4741表示正常酵母,R-2表示變異酵母,箭頭所指處為X號染色體條帶;
圖4所示為酵母R-2基因組測序深度圖;
圖5所示為篩選標簽培養基篩選染色體結構修復的菌株結果;其中,1-3表示不同四分體下的4個孢子,YPD為正常培養基,-URA為缺URA的篩選培養基,-LEU為缺LEU培養基,圖中圓圈選定的孢子為修復后的菌株,即標號1的四分體的第4個孢子;
圖6所示為PCR驗證染色體結構變異修復正常的凝膠圖;其中,BY4741表示正常酵母,R-2表示變異酵母,synX表示修復后的酵母。
具體實施方式
本發明公開了一種修復酵母染色體結構異常的方法,本領域技術人員可以借鑒本文內容,適當改進工藝參數實現。特別需要指出的是,所有類似的替換和改動對本領域技術人員來說是顯而易見的,它們都被視為包括在本發明。本發明所述方法已經通過較佳實施例進行了描述,相關人員明顯能在不脫離本發明內容、精神和范圍內對本文所述的方法進行改動或適當變更與組合,來實現和應用本發明技術。
根據本發明記載的修復方法,其可以概括為如下:
(1)確認染色體結構變異的區域;(2)在染色體結構變異出現的末端位置插入篩選標記基因;(3)在染色體結構正常的單倍體酵母對應位置的鏈交換區域插入另一個篩選標記基因;(4)染色體變異的酵母和染色體正常的酵母交配形成二倍體;(5)減數分裂生孢;(6)孢子拆分,并進行篩選基因的表型驗證。
以下就本發明所提供的一種修復酵母染色體結構異常的方法做進一步說明。
實施例1:修復染色體結構異常的全合成型染色體釀酒酵母R-2
脈沖場凝膠電泳對釀酒酵母R-2菌進行核型分析,觀察圖3可以看到菌株R-2和野生型對照菌株BY4741相比,十號染色體對應的條帶明顯向上發生了遷移。在全基因組測序分析菌株R-2后,通過圖4測序深度圖發現有兩處區域有大片段復制重復。一處主要發生在megachunk C區域,涉及序列長度為101kb,為連續發生的三拷貝的復制。第二處主要發生在megachunk E區域,涉及序列長度為147kb,為二拷貝和四拷貝交錯排布的復制。綜合計算R-2菌株十號染色體的長度比野生型長度長171kb。針對重復區域特異性的連接處位置的序列設計引物,連接處設計擴增大小分別為1kb,500bp兩對引物。從結果可以看出只有發生大片段重復的菌株才能擴增出大小正確的條帶,而對照BY4741并無條帶擴增。將PCR產物送測,可以得到斷裂位點的明確序列。根據全基因組測序拼接結果及連接處PCR產物測序結果分析得到染色體結構異常的具體位置信息。
利用酵母細胞減數分裂時非姐妹染色單體的同源重組實現結構異常的染色體和結構正常的染色體之間的鏈交換(圖2所示示意圖)。在單倍體酵母菌株R-2中出現染色體結構變異的末端位置插入URA基因。在染色體結構正常的單倍體半合成型染色體酵母BY4742中(與染色體結構變異的單倍體R-2不同的交配型),將LEU基因插入到與染色體結構異常區域對應的鏈交換區域。將上述兩個不同交配型的菌株R-2和BY4742交配形成二倍體酵母,在生孢培養基中培養二倍體菌株4到7天使其生孢。
二倍體酵母細胞液體生孢的流程如下:
1.預生孢。在30℃YPD液體培養基中培養二倍體酵母細胞至OD600值達到4~8。
2.漂洗。2000g室溫離心2min將預生孢的二倍體細胞沉淀,移除上清液并用超純水重懸細胞。重復上述漂洗過程兩次。
3.生孢。用超純水稀釋50X生孢液到1X濃度。添加需要補充的氨基酸(依據二倍體酵母細胞的營養缺陷情況)。每毫升的1X生孢液需要額外添加3μl10%的酵母提取物。混合均勻。把漂洗干凈的預生孢二倍體酵母加入混合好的生孢培養基內使其OD600值大于1。在25℃下培養3-10天。通過顯微觀察是否有四分體孢子出現來檢驗是否生孢成功。
YPD培養基:1%酵母提取物,2%蛋白胨,2%葡萄糖。
50X預生孢培養基:50%醋酸鉀,0.25%二水乙酸鋅加超純水溶解。過濾除菌,室溫保存。
10%酵母提取物:10%酵母提取物溶解于超純水。4℃保存。
使用酵母顯微操作儀對生孢的四分體細胞進行拆分,拆分步驟如下:
1.吸取50uL生孢液,2000g離心2分鐘。
2.去除上清夜,重懸細胞在25uL的0.5mg/mL的裂解酶緩沖液中(溶劑為1M山梨醇)。
3.在37℃條件下孵育10分鐘。
4.用1M山梨醇稀釋至300uL,置于冰上。
5.利用酵母顯微拆分儀,將生孢成功的四分體細胞打散并挑取到對應的平板位置。需要至少拆分十個四分體細胞。
拆分的單倍體細胞按照四分體的來源整齊排布在YPD平板上培養2天,之后將生長的單倍體孢子平板翻印到SC-URA和SC-LEU的平板來篩選在設定區間發生鏈交換的孢子。挑取既不能在SC-URA平板上生長也不能在SC-LEU平板上生長的單倍體孢子,只有在設定區間發生了鏈交換才會引起兩個標記基因位置的變換。通過圖5可知,來自同一個四分體的孢子在SC-URA,SC-LEU平板上都呈現2:2分布,完全符合鏈交換后的比例分布,其中四分體1的第4個孢子(被圓圈標注)既不能在SC-URA平板上生長也不能在SC-LEU平板上生長,其即為修復成功的菌株,命名為synX。
挑取通過表型驗證的單倍體孢子synX,利用PCR驗證染色體結構異常是否被修復,從圖6 PCR驗證膠圖結果可以看出,R-2菌株染色體特異性的異常結構不能在修復后的菌株synX中擴增出來,表明染色體結構異常被修復成功。
注:菌株R-2對應的野生型染色體酵母是BY4741,兩者交配型相同,區別在于一個是全合成型染色體酵母,一個是野生型染色體;BY4742與BY4741僅為交配型不同,本實施中的BY4742為半合成型染色體酵母,存在的合成型染色體位置對應于R-2中出現染色體結構異常的區域,其他位置染色體均為野生型染色體,與BY4741相同。
以上所述僅是本發明的優選實施方式,應當指出,對于本技術領域的普通技術人員來說,在不脫離本發明原理的前提下,還可以做出若干改進和潤飾,這些改進和潤飾也應視為本發明的保護范圍。
- 還沒有人留言評論。精彩留言會獲得點贊!