一種改性聚丙烯酰胺及其制備方法和應用與流程
本發明屬于吸附技術領域,涉及一種改性聚丙烯酰胺及其制備方法和應用。
背景技術:
在我國,企業每年排放的工業污水約為199億噸,其中染料廢水約為70億噸,因此染料廢水是工業廢水的主要來源。隨著印染行業技術的發展,染料廢水呈現出可生化性逐漸變差的趨勢,且染料廢水還具有色度深、cod值高、毒性強、濃度高的特點,使其逐漸成為我國廢水處理的主要難點。目前,處理染料廢水常用的方法主要有生物降解、氧化、膜技術和吸附。國內外主要采用生物法和混凝法處理染料廢水,但目前處理染料廢水具有難生化降解性,僅靠生物處理方法難以使染料廢水達標排放,而吸附法則有較好的處理效果,因此,低污染,低成本,高選擇性的吸附劑成為了治理染料廢水的有效手段。目前工業上常采用的吸附劑為活性炭。活性炭造價高,普遍用于工業廢水的深度處理。
聚丙烯酰胺(以下簡稱pam)是重要的高分子聚合物之一,性質為水溶性、高分子,是通過丙烯酰胺在特定條件下均聚或與其他單體共聚而成,質量分數大于50%的線性的水溶性的化學物品的總稱。依據物質的性狀pam產品主要有三種類型:乳液、水溶液膠體、粉狀;依據物質的離子特性分為陽離子型(cpam)、陰離子型(apam)、非離子型(npam)及兩性型,四種類型的聚合物有的是均聚物,也有的是共聚物。
陰離子聚丙烯酰胺是常用的高分子絮凝劑,在污水處理、化工、石油等領域被廣泛應用。其特點是絮凝性,粘合性。因此也是最受人們關注的,其重要性是生產成本遠低于陽離子高分子絮凝劑和兩性高分子絮凝劑。
陰離子型聚丙烯酰胺的合成方法主要有水解法,共聚法。水解法:均聚后水解法、均聚共水解法。共聚法:單體共聚法、接枝共聚法、水溶液聚合、反相乳液聚合及反相懸浮聚合。當前陰離子型聚丙烯酰胺的生產大多采用的是共聚法。
沈雷(陰離子聚丙烯酰胺絮凝劑(apam)的制備及應用[d].重慶大學,2011)采用的水溶液共聚法,在無機引發劑過硫酸銨和亞硫酸氫鈉做引發劑的條件下,聚合丙烯酰胺單體和氫氧化鈉得到陰離子聚丙烯酰胺(apam),實驗研究合成了分子量為960萬,且殘留單體含量為0.072%;與此同時獲得另兩種高分子聚合物apam分子量分別為872萬和883萬,殘留單體物質的含量為0.050%和0.043%。
通過上述對中國水資源和水污染的現狀描述,以及聚丙烯酰胺的合成以及改性和應用方面的陳述,陰離子聚丙烯酰胺絮凝劑的應用在水處理技術中發揮著重要作用,而我國在陰離子聚丙烯酰胺絮凝劑在其制備的種類生產上目前還處于一般的水平。
改性聚丙烯酰胺可以作為一種親水性聚合物,當前已應用于有機工業,生物醫學、水的凈化處理、食品工業、重金屬回收、懸浮物的絮沉。此聚合物具有多種功能官能團,如羧基、氨基、羥基或磺酸基等。利用胺甲基化反應(mannich反應)即羥甲基反應和接枝反應在陰離子聚丙烯酰胺主鏈上引入含氨基的基團,當引入的基團為氨基或者其他基團時,就可以提升物質本身的功能性,拓展改性物質的在其他方面的應用。
技術實現要素:
本發明的目的在于解決現有技術中存在的上述技術和成本問題,提供一種改性聚丙烯酰胺及其制備方法和應用,以聚丙烯酰胺為母體,與配體(2-氨基-5-巰基-1,3,4-噻二唑)進行合成反應,能夠獲得具有較高功能基轉化率的改性聚丙烯酰胺,該改性聚丙烯酰胺對堿性品紅具有良好的選擇性吸附性能。
為解決上述技術難題,本發明采用以下技術方案:
一種改性聚丙烯酰胺,包含如式(ⅰ)所示的重復單元:
所述改性聚丙烯酰胺的特性粘度為700~800mg/ml。
母體聚丙烯酰胺是能吸附微粒官能團的線性高分子化合物,能將微粒吸附在一起形成團狀物聚丙烯酰胺長碳鏈,在微粒之間起著架橋作用。聚丙烯酰胺可高分子骨架通過與試劑發生反應連接功能基團,從而制備可分離富集的功能材料。配體2-氨基-5-巰基-1,3,4-噻二唑中具有多個氮原子,
能提供孤對電子,這樣的化學結構能更好地螯合堿性品紅。
所述改性聚丙烯酰胺的制備方法,包括:以具有如(ⅱ)所示重復單元的陰離子聚丙烯酰胺為母體,與甲醛發生羥甲基反應,再與結構如(ⅲ)所示的配體發生接枝反應,得到具有所述改性聚丙烯酰胺;
所述制備方法具體包括:
(1)將陰離子聚丙烯酰胺溶于水中,加入甲醛水溶液進行羥甲基反應,得混合物;
(2)在步驟(1)得到的混合物中加入所述配體進行接枝反應,得聚合物;
(3)將步驟(2)得到的聚合物浸泡在溶劑中,經水洗滌后,冷凍干燥得所述改性聚丙烯酰胺。
優選地,步驟(1)中,將陰離子聚丙烯酰胺溶于水中,所得陰離子聚丙烯酰胺水溶液的質量分數為0.5~5%。
優選地,步驟(1)中,所述甲醛水溶液的質量分數為20~50%。
優選地,步驟(1)中,所述陰離子聚丙烯酰胺與甲醛水溶液的投加比例為1g:0.1~8ml,在該比例范圍內,甲醛的利用率隨甲醛投加比例的增加先增加后減小,由于陰離子聚丙烯酰胺與甲醛反應生成羥甲基聚丙烯酰胺中間體,再與配體進行接枝反應,若甲醛含量過高,易發生交聯,會降低產品的穩定性;進一步優選,所述陰離子聚丙烯酰胺與甲醛水溶液的投加比例為1g:2~8ml;最優地,所述陰離子聚丙烯酰胺與甲醛水溶液的投加比例為1g:3ml,此時甲醛的利用率最高,達到87.6%。
優選地,步驟(1)中,所述羥甲基反應的溫度為25~35℃。
優選地,步驟(1)中,所述羥甲基反應的時間為2~5h。
優選地,步驟(2)中,所述配體與步驟(1)中陰離子聚丙烯酰胺的質量比為1:0.3~1.1,隨著配體投加比例的增加,所得改性陰離子聚丙烯酰胺的分子量先增加,然后增加趨勢趨于平緩。由于隨著反應時間的增加,聚合接枝反應不斷進行,聚合物的分子鏈不斷的加長,使聚合物的分子量不斷的增加,而當反應進行到一定時間時,由于主鏈上可接枝的分子鏈數量有限,總分子量停止增加。
優選地,步驟(2)中,所述接枝反應的溫度為50~100℃,制備的改性聚丙烯酰胺的分子量是隨溫度的升高而增大,最后趨于平衡;由于接枝反應溫度的升高可以提高溶液中的配體向母體(陰離子聚丙烯酰胺)主鏈上的反應活性位點擴散的速率,并且增大了活性位點配體的濃度,促進聚合反應的進行。
進一步優選,所述接枝反應的溫度為70~100℃。在該溫度范圍內,所得改性聚丙烯酰胺的分子量最大。
優選地,在步驟(2)中,所述接枝反應的ph為6~8,所得改性聚丙烯酰胺的分子量隨著ph的升高先增大后減小;進一步優選,所述接枝反應的ph為7.5,該ph下,反應環境的游離的氫離子和氫氧根離子相互作用沒有干擾聚合反應。
優選地,步驟(2)中,所述接枝反應的時間為8~15h。
優選地,步驟(3)中,所述溶劑為質量分數為20~50%的甲醛水溶液。
優選地,步驟(3)中,所述冷凍干燥的溫度為-60~-40℃。
本發明還提供了一種所述改性聚丙烯酰胺在吸附堿性品紅中的應用。
與現有技術相比,采用本發明的方法制備的改性聚丙烯酰胺,具有如下優點:原料來源廣泛,價格低廉;
1、本發明制備方法簡單,操作方便,產率高;
2、將陰離子聚丙烯酰胺進行改性,使其具有機械強度高,熱穩定性好的特點;
3、反應溶劑以及反應物均綠色無毒,節約成本,保護環境,減少了二次污染;
4、本發明改性聚丙烯酰胺是可分離富集的功能材料,含有較多的功能基團,對堿性品紅有較好的吸附性;
5、本發明改性聚丙烯酰胺的氮原子較多,含氮量高,提供孤對電子多,對堿性品紅有較好的吸附效果;
6、本發明提供的反應路線簡單,合成方法操作方便,只需要母體與甲醛反應生成羥甲基化聚丙烯酰胺中間體后然后與配體發生接枝反應兩步即可,條件容易達到,容易實現批量生產及自動化控制,具有良好的應用前景
附圖說明
圖1為實施例1制備的改性聚丙烯酰胺的紅外譜圖;
圖2為apam和實施例1制備的eamtdp的熱失重曲線;
圖3為apam和實施例1制備的eamtdp的熱失重速率分析圖;
圖4為apam和實施例1制備的eamtdp的掃描電鏡圖,其中,(a)為apam的掃描電鏡圖,(b)為eamtdp的掃描電鏡圖;
圖5為體系ph對堿性品紅去除率的影響結果圖;
圖6為吸附溫度對堿性品紅去除率的影響結果圖。
具體實施方式
下面結合具體實施例來進一步描繪本發明,但本發明的內容并不限于此。
本發明中,測定聚合物粘度的方法如下:
準確稱取0.1g改性聚丙烯酰胺樣品,放入盛有50ml去離子水的燒杯中,在磁力攪拌器上加熱攪拌數分鐘,之后關掉加熱開關,然后取下燒杯,冷卻至室溫,再將溶液轉移至100ml干凈干燥的容量瓶中,加入一定量的去離子水進行定容,備用。
實驗的測量方法和具體測量操作依據國家標準gb12005.1-89“聚丙烯酰胺特性粘度測定方法”;實驗測試高分子聚合物的特性粘數[η]是依據國家標準gbpt12005.10-92“聚丙烯酰胺分子量測定——粘度法”,計算相關的數據得到高分子聚合物的粘均相對分子質量。
聚合物的相對分子質量可根據按式(1)(2)(3)計算:
ηr=t/to(1)
ηr——相對粘度;
t——試樣溶液的流經時間,s;
to——1.00mol/l氯化鈉溶液的流經時間,s。
c=ms/v(2)
由求得的ηr查表得相應的[η]·c值除以試樣濃度c即得特性粘度[η]。
式中:
[η]——特性粘度,ml/g;
c——試樣溶液的濃度,g/ml;
m——試樣質量,g;
s——試樣固含量,g;
v——制備的試樣溶液體積,ml
本測試條件的mark-houwink公式如下:
[η]=kma(3)
式中:
[η]——溶液的特性粘度,ml/g;
k——4.75×10-3,ml/g;
m——相對分子質量
a——0.80。
實施例1
一種改性聚丙烯酰胺的制備方法,依次進行以下步驟:
(1)準確稱取陰離子聚丙烯酰胺(apam)1.0g,配成質量分數約為1%的水溶液,在常溫下充分溶解,加入調節ph至7.5,加入3ml質量分數為36.69%的甲醛溶液,在常溫下反應3h,得混合物;通過測定混合物中的剩余甲醛的量,計算分析得甲醛的利用率為87.6%。
(2)在步驟(1)所得混合物中加入0.80g(0.006mol)配體2-氨基-5-巰基-1,3,4-噻二唑(eamtd),并在70℃下,ph為7.5,氮氣保護下加熱攪拌反應10h,攪拌速度為300rpm/min,得聚合物;
(3)將步驟(2)所得的聚合物用質量分數為36.69%的甲醛水溶液浸泡,然后用蒸餾水洗滌數次;
將上述沖洗后的聚合物放入-50℃溫度下冷凍至恒重,得到所述改性聚丙烯酰胺(eamtdp)。
經檢測,所得改性聚丙烯酰胺的特性粘度為786.49mg/ml,計算得所述改性聚丙烯酰胺的相對分子質量3.34×106。
所得改性聚丙烯酰胺的紅外譜圖如圖1所示,1600cm-1處為c=n彎曲振動吸收峰,699cm-1處為c-s伸縮振動吸收峰,1400cm-1為c-n伸縮振動吸收峰,3350,3180cm-1處為n-h伸縮振動吸收峰;比較改性聚丙烯酰胺(eamtdp)、陰離子聚丙烯酰胺(apam)的紅外光譜曲線可知它們之間有著較大的差異,尤其是在改性譜圖中699cm-1,1600cm-1處有強吸收峰出現,并且1400cm-1,3000-3500cm-1之間的峰值明顯加強,由此表明合成后的物質中增加了c-s,c=n,也證明合成的物質具有豐富的氨基基團。
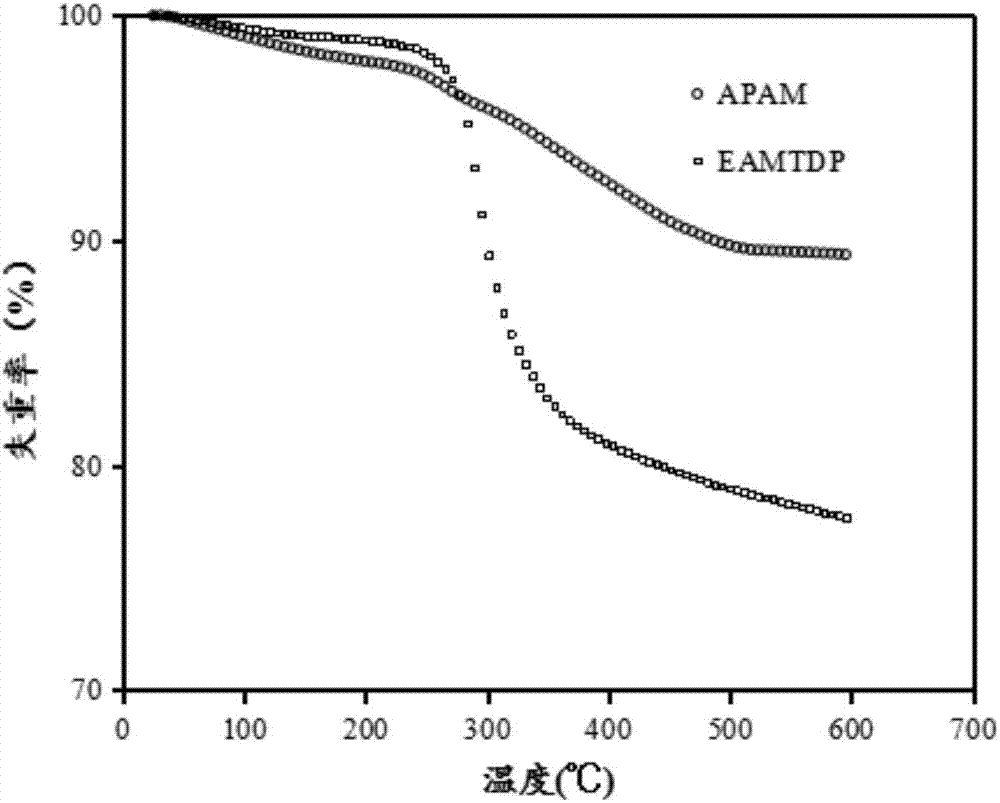
聚丙烯酰胺(apam)和改性聚丙烯酰胺(eamtdp)的熱失重曲線如圖2所示,聚丙烯酰胺(apam)和改性聚丙烯酰胺(eamtdp)的熱失重速率分析如圖3所示。
當溫度升至100℃左右時,聚丙烯酰胺的熱失重率開始大幅度變化,在500℃之后改變的不明顯。改性聚丙烯酰胺的熱分解過程則分為三個階段。
apam:第一階段起始于25℃,終止于145℃左右,熱失重量為1.59%,熱失重速率為0.000217%s-1;第二階段是主鏈上支鏈鍵的斷裂coo-,conh2中的c=o,c-o,c-n,n-h發生斷裂,結果是聚丙烯酰胺質量明顯改變,起始于175~211℃,終止于475℃左右;第三階段是主鏈在高溫度下分解。
eamtdp:第一階段起始于25℃,終止于139℃左右,失重率為0.81%,熱失重率速率為0.00011%s-1;第二階段是原有的主鏈上的支鏈和接枝上的支鏈的裂解即coo-和
從圖3的熱失重速率來看,在apam的55、271℃,343℃;eamtdp的55℃、199℃、295℃出現熱速率峰值表明在對應溫度值下發生官能團的斷裂結果是重量在短時間內變化很大。eamtdp在高溫下穩定性變差,但在常溫下是穩定的。
聚丙烯酰胺(apam)和改性聚丙烯酰胺(eamtdp)的掃描電鏡圖如圖4所示,其中,圖4(a)為聚丙烯酰胺(apam)的掃描電鏡圖,圖4(b)為改性聚丙烯酰胺(eamtdp)的掃描電鏡圖,可以看出,apam和eamtdp的結構明顯不同,二者顆粒大小明顯改變,eamtdp的顆粒明顯小于apam,二者的表面積也隨之改變。因此得出合成前后的物質apam和eamtdp的結構不同。
實施例2
一種改性聚丙烯酰胺的制備方法,依次進行以下步驟:
(1)準確稱取陰離子聚丙烯酰胺1.0g,配成質量分數約為1%的水溶液,在常溫下充分溶解,加入調節ph至7.5,加入6ml質量分數為35%的甲醛溶液,反應2h,得混合物;通過測定混合物中的剩余甲醛的量,計算分析得甲醛的利用率為85.3%。
(2)在步驟(1)所得混合物中加入0.37g(0.005mol)配體2-氨基-5-巰基-1,3,4-噻二唑(eamtd),并在70℃下,ph為7.5,氮氣保護下加熱攪拌反應8h,攪拌速度為300rpm/min,得聚合物;
(3)將步驟(2)所得的聚合物用質量分數為35%的甲醛水溶液浸泡,然后用蒸餾水洗滌數次;
將上述沖洗后的聚合物放入-60℃溫度下冷凍至恒重,得到所述改性聚丙烯酰胺。
經檢測計算,所得改性聚丙烯酰胺的相對分子質量3.28×106。
實施例3
一種改性聚丙烯酰胺的制備方法,依次進行以下步驟:
(1)準確稱取陰離子聚丙烯酰胺1.0g,配成質量分數約為1%的水溶液,在常溫下充分溶解,加入調節ph至7,加入8ml質量分數為25%的甲醛溶液,反應5h,得混合物;通過測定混合物中的剩余甲醛的量,計算分析得甲醛的利用率為83.6%。
(2)在步驟(1)所得混合物中加入0.53g(0.004mol)配體2-氨基-5-巰基-1,3,4-噻二唑(eamtd),并在70℃下,ph為7.5,氮氣保護下加熱攪拌反應13h,攪拌速度為300rpm/min,得聚合物;
(3)將步驟(2)所得的聚合物用質量分數為25%甲醛水溶液浸泡,然后用蒸餾水洗滌數次;
將上述沖洗后的聚合物放入-40℃溫度下冷凍至恒重,得到所述改性聚丙烯酰胺。
經檢測計算,所得改性聚丙烯酰胺的相對分子質量3.25×106。
實施例4
一種改性聚丙烯酰胺的制備方法,依次進行以下步驟:
(1)準確稱取陰離子聚丙烯酰胺1.0g,配成質量分數約為1%的水溶液,在常溫下充分溶解,加入調節ph至7.5,加入2ml質量分數為45%的甲醛溶液,反應2h,得混合物;通過測定混合物中的剩余甲醛的量,計算分析得甲醛的利用率為83.4%。
(2)在步驟(1)所得混合物中加入0.40g(0.003mol)配體2-氨基-5-巰基-1,3,4-噻二唑(eamtd),并在70℃下,ph為7.5,氮氣保護下加熱攪拌反應8h,攪拌速度為300rpm/min,得聚合物;
(3)將步驟(2)所得的聚合物用質量分數為45%的甲醛水溶液浸泡,然后用蒸餾水洗滌數次;
將上述沖洗后的聚合物放入-60℃溫度下冷凍至恒重,得到所述改性聚丙烯酰胺。
經檢測計算,所得改性聚丙烯酰胺的相對分子質量3.14×106。
實施例5
一種改性聚丙烯酰胺的制備方法,依次進行以下步驟:
(1)準確稱取陰離子聚丙烯酰胺1.0g,配成質量分數約為1%的水溶液,在常溫下充分溶解,加入調節ph至7.5,加入6ml質量分數為36.69%的甲醛溶液,反應2h,得混合物;通過測定混合物中的剩余甲醛的量,計算分析得甲醛的利用率為84.1%。
(2)在步驟(1)所得混合物中加入1.07g(0.008mol)配體2-氨基-5-巰基-1,3,4-噻二唑(eamtd),并在70℃下,ph為7.5,氮氣保護下加熱攪拌反應8h,攪拌速度為300rpm/min,得聚合物;
(3)將步驟(2)所得的聚合物用質量分數為36.69%甲醛水溶液浸泡,然后用蒸餾水洗滌數次;
將上述沖洗后的聚合物放入-60℃溫度下冷凍至恒重,得到所述改性聚丙烯酰胺。
經檢測計算,所得改性聚丙烯酰胺的相對分子質量3.34×106。
實施例6
一種改性聚丙烯酰胺的制備方法,依次進行以下步驟:
(1)準確稱取陰離子聚丙烯酰胺1.0g,配成質量分數約為3%的水溶液,在常溫下充分溶解,加入調節ph至7.5,加入8ml質量分數為50%的甲醛溶液,反應5h,得混合物;通過測定混合物中的剩余甲醛的量,計算分析得甲醛的利用率為87.3%。
(2)在步驟(1)所得混合物中加入0.80g(0.006mol)配體2-氨基-5-巰基-1,3,4-噻二唑(eamtd),并在50℃下,ph為7.5,氮氣保護下加熱攪拌反應15h,攪拌速度為300rpm/min,得聚合物;
(3)將步驟(2)所得的聚合物用質量分數為50%的甲醛水溶液浸泡,然后用蒸餾水洗滌數次;
將上述沖洗后的聚合物放入-50℃溫度下冷凍至恒重,得到所述改性聚丙烯酰胺。
經檢測計算,所得改性聚丙烯酰胺的相對分子質量3.31×106。
實施例7
一種改性聚丙烯酰胺的制備方法,依次進行以下步驟:
(1)準確稱取陰離子聚丙烯酰胺1.0g,配成質量分數約為3%的水溶液,在常溫下充分溶解,加入調節ph至7.5,加入8ml質量分數為50%的甲醛溶液,反應5h,得混合物;通過測定混合物中的剩余甲醛的量,計算分析得甲醛的利用率為87.3%。
(2)在步驟(1)所得混合物中加入0.80g(0.006mol)配體2-氨基-5-巰基-1,3,4-噻二唑(eamtd),并在60℃下,ph為7.5,氮氣保護下加熱攪拌反應13h,攪拌速度為300rpm/min,得聚合物;
(3)將步驟(2)所得的聚合物用質量分數為50%的甲醛水溶液浸泡,然后用蒸餾水洗滌數次;
將上述沖洗后的聚合物放入-50℃溫度下冷凍至恒重,得到所述改性聚丙烯酰胺。
經檢測計算,所得改性聚丙烯酰胺的相對分子質量3.29×106。
實施例8
一種改性聚丙烯酰胺的制備方法,依次進行以下步驟:
(1)準確稱取陰離子聚丙烯酰胺1.0g,配成質量分數約為1%的水溶液,在常溫下充分溶解,加入調節ph至7.5,加入3ml質量分數為36.69%的甲醛溶液,反應3h,得混合物;通過測定混合物中的剩余甲醛的量,計算分析得甲醛的利用率為87.6%。
(2)在步驟(1)所得混合物中加入0.80g(0.006mol)配體2-氨基-5-巰基-1,3,4-噻二唑(eamtd),并在70℃下,ph為8,氮氣保護下加熱攪拌反應8h,攪拌速度為300rpm/min,得聚合物;
(3)將步驟(2)所得的聚合物用質量分數為36.69%的甲醛水溶液浸泡,然后用蒸餾水洗滌數次;
將上述沖洗后的聚合物放入-50℃溫度下冷凍至恒重,得到所述改性聚丙烯酰胺。
經檢測,所得改性聚丙烯酰胺的相對分子質量3.25×106。
實施例9
一種改性聚丙烯酰胺的制備方法,依次進行以下步驟:
(1)準確稱取陰離子聚丙烯酰胺1.0g,配成質量分數約為1%的水溶液,在常溫下充分溶解,加入調節ph至7.5,加入3ml質量分數為36.69%的甲醛溶液,反應3h,得混合物;通過測定混合物中的剩余甲醛的量,計算分析得甲醛的利用率為87.6%。
(2)在步驟(1)所得混合物中加入0.80g(0.006mol)配體2-氨基-5-巰基-1,3,4-噻二唑(eamtd),并在70℃下,ph為6,氮氣保護下加熱攪拌反應8h,攪拌速度為300rpm/min,得聚合物;
(3)將步驟(2)所得的聚合物用質量分數為36.69%的甲醛水溶液浸泡,然后用蒸餾水洗滌數次;
將上述沖洗后的聚合物放入-50℃溫度下冷凍至恒重,得到所述改性聚丙烯酰胺。
經檢測,所得改性聚丙烯酰胺的相對分子質量3.22×106。
實施例10
稱取10.0mg實施例1所得的改性聚丙烯酰胺(eamtdp),放置于干凈干燥的100ml碘量瓶中,之后加入一定體積的去離子水,使改性聚丙烯酰胺充分溶解,然后加入30ml的濃度為200mg/l的堿性品紅標準液,調好體系ph=7.5,在30℃的恒溫振蕩器中并且以一定的300rpm/min的轉速振搖至平衡進行吸附試驗。吸附結束后,經過高速離心15min后,取距液面30mm處的上清液在堿性品紅最大吸收波長542nm處測定溶液的吸光度,分析溶液的水相中殘余堿性品紅的濃度。然后用以下公式計算出堿性品紅的去除率:
d(%)=(co-ce)/co
式中:ce為溶液平衡后的堿性品紅溶液的濃度(mg/l);co則為吸附前的堿性品紅溶液的濃度(mg/ml)。
推算出改性聚丙烯酰胺(eamtdp)對堿性品紅的去除率為83.79%。
實施例11~14
重復實施例10的操作,區別僅在于體系的ph值分別調節至5.0、6.0、7.0、8.0,以研究體系ph對堿性品紅去除率的影響,結果如圖5所示。
在改性聚丙烯酰胺吸附堿性品紅過程中,溶液初始ph是吸附實驗重要的影響因素之一。實驗反應溶液中氫離子極易被吸附,并且氫氧根離子也會影響吸附位點,因此,探究ph對堿性品紅的吸附有重要的意義。
根據圖5可知,體系初始ph值的不斷增大改性聚丙烯酰胺顆粒對堿性品紅吸附量的值也增大。當ph值由5增加到6后脫色率緩慢增長。在中性條件下,溶液中的氫離子和氫氧根離子濃度相等達到平衡不會影響吸附的進行,使得吸附物質對堿性品紅的吸附能力上升。當溶液初始ph達到7.5時脫色率最高。
實施例15~18
重復實施例10的操作,區別僅在于體系的溫度改為15℃、20℃、25℃、35℃,以研究吸附溫度對堿性品紅去除率的影響,結果如圖6所示。
由圖6中曲線中可以得出,反應進行到30℃后,吸附實驗基本達到平衡。因此30℃被定為最佳吸附溫度。在30℃之前溫度的改變會顯著影響脫色率的結果,在30℃后影響不大,原因在于溫度升高改性聚丙烯酰胺吸附劑的官能團和吸附位點比較活躍,分子作用強烈,大大促進了堿性品紅分子由邊界層逐步擴散到吸附劑內表而及其內部。
上述實施例不以任何方式限制本發明,凡是采用等同替換或等效變換的方式獲得的技術方案均落在本發明的保護范圍內。
- 還沒有人留言評論。精彩留言會獲得點贊!