一種以三嗪和苯并咪唑為核心的有機化合物及其在有機電致發光器件上的應用的制作方法
本發明涉及半導體
技術領域:
,尤其是涉及一種以三嗪和苯并咪唑為核心的有機化合物及其在有機電致發光器件上的應用。
背景技術:
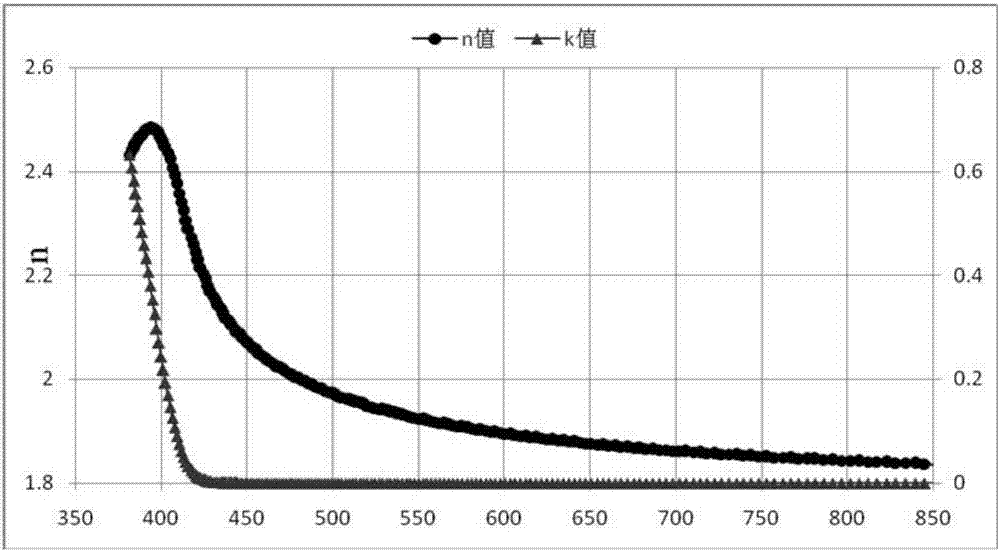
:有機電致發光(oled:organiclightemissiondiodes)器件技術既可以用來制造新型顯示產品,也可以用于制作新型照明產品,有望替代現有的液晶顯示和熒光燈照明,應用前景十分廣泛。oled發光器件猶如三明治的結構,包括電極材料膜層以及夾在不同電極膜層之間的有機功能材料,各種不同功能材料根據用途相互疊加在一起共同組成oled發光器件。oled發光器件作為電流器件,當對其兩端電極施加電壓,并通過電場作用有機層功能材料膜層中的正負電荷時,正負電荷進一步在發光層中復合,即產生oled電致發光。當前,oled顯示技術已經在智能手機,平板電腦等領域獲得應用,進一步還將向電視等大尺寸應用領域擴展。但是,由于oled的外量子效率和內量子效率之間存在巨大差距,極大地制約了oled的發展。因此,如何提高oled的光取出效率成為研究熱點。ito薄膜和玻璃襯底的界面以及玻璃襯底和空氣的界面處會發生全反射,出射到oled器件前向外部空間的光約占有機材料薄膜el總量的20%,其余約80%的光主要以導波形式限制在有機材料薄膜、ito薄膜和玻璃襯底中。可見常規oled器件的出光效率較低(約為20%),這嚴重制約了oled的發展和應用。如何減少oled器件中的全反射效應、提高光耦合到器件前向外部空間的比例(出光效率)引起人們的廣泛關注。目前,實現提高oled外量子效率的一類重要方法是在基底出光表面形成如褶皺、光子晶體、微透鏡陳列(mla)和添加表面覆蓋層等結構。前兩種結構會影響oled的輻射光譜角度分布,第三種結構制作工藝復雜,使用表面覆蓋層工藝簡單,發光效率提高30%以上,尤為人們關注。根據光學原理,當光透射過折射率為n1的物質到折射率為n2的物質時(n1>n2),只有在arcsin(n2/n1)的角度內才能入射到折射率為n2的物質里,吸收率b可以用以下的公式計算:設n1=n一般oled有機材料=1.70,n2=n玻璃=1.46,則2b=0.49。假設向外傳播的光全部被金屬電極反射,則只有51%的光能被高折射率的有機膜和ito層所波導,同樣可以計算光從玻璃基底射出到空氣時的透過率。因此從有機層發出的光射出器件的外部時,只有約17%的光能被人們所看見。因此,針對目前oled器件光取出效率低的現狀,需要在器件結構中增加一層cpl層,即光提取材料,根據光學吸收、折射原理,此表面覆蓋層材料的折射率應該越高越好。目前對oled發光器件提高性能的研究包括:降低器件的驅動電壓、提高器件的發光效率、提高器件的使用壽命等。為了實現oled器件的性能的不斷提升,不但需要從oled器件結構和制作工藝的創新,更需要oled光電功能材料不斷研究和創新,創制出更高性能的oled功能材料。技術實現要素:針對現有技術存在的上述問題,本申請人提供了一種以三嗪和苯并咪唑為核心的有機化合物及其在有機電致發光器件上的應用。本發明化合物含有三嗪及苯并咪唑結構,具有較高的玻璃化溫度和分子熱穩定性,在可見光領域吸收低、折射率高,在應用于oled器件的cpl層后,可有效提升oled器件的光取出效率;并且由于三嗪和苯并咪唑具有深的homo能級,寬的禁帶(eg)能級,可作為oled器件的空穴阻擋/電子傳輸層材料,阻擋空穴從發光層傳遞至電子層一側,提高空穴和電子在發光層中的復合度,從而提升oled器件的發光效率和使用壽命。本發明的技術方案如下:本申請人提供了一種以三嗪和苯并咪唑為核心的有機化合物,所述有機化合物的結構如通式(1)所示:通式(1)中,ar1表示為c1-10直鏈或支鏈烷基、鹵素原子、氕、氘、氚原子取代或未取代的苯基、萘基、二聯苯基、三聯苯基、蒽基、二苯并呋喃、二苯并噻吩、9,9-二甲基芴或9-苯基咔唑;ar2、ar3分別獨立的表示為c1-10直鏈或支鏈烷基、鹵素原子、氕、氘、氚原子取代或未取代的苯基、萘基、二聯苯基、三聯苯基、蒽基、吡啶基的一種;ar2、ar3還分別獨立的表示為單鍵;ar1、ar2、ar3可以相同或不同;r1、r2分別獨立的表示為通式(2)、通式(3)或通式(4)所示結構;其中,ar4、ar5、ar6、ar7分別獨立的表示為c1-10直鏈或支鏈烷基、鹵素原子取代或未取代的苯基、萘基、二聯苯基、三聯苯基、吡啶基的一種;r1、r2可以相同或不同。優選的,所述有機化合物的具體結構式為:中的任意一種。本申請人還提供了所述的有機化合物的制備方法,制備過程中發生的反應方程式為:第一步反應方程式具體反應過程為:第一步:氮氣氛圍下,稱取原料2,4,6-三氯-1,3,5-三嗪溶解于四氫呋喃中,再將ar1的硼酸化合物及四(三苯基膦)鈀加入,攪拌混合物,再加入飽和碳酸鉀水溶液,將上述反應物的混合溶液于反應溫度70-90℃下加熱回流10-20小時;反應結束后,冷卻、混合液用二氯甲烷萃取,萃取物用無水硫酸鈉干燥處理,且在減壓下濃縮,將濃縮固體過硅膠柱純化,得到化合物中間體i;所述2,4,6-三氯-1,3,5-三嗪與ar1-b(oh)2的摩爾比為1:1.0~1.5,pd(pph3)4與2,4,6-三氯-1,3,5-三嗪的摩爾比為0.005~0.05:1,k2co3與2,4,6-三氯-1,3,5-三嗪的摩爾比為1.0~2.0:1,thf用量為:2,4,6-三氯-1,3,5-三嗪:thf=1g:10~20ml;第二步反應方程式第二步:氮氣氛圍下,稱取中間體i溶解于n,n-二甲基甲酰胺中,再將及醋酸鈀加入,攪拌混合物,再加入磷酸鉀水溶液,將上述反應物的混合溶液于反應溫度120-150℃下加熱回流10-24小時;反應結束后,冷卻加水、將混合物過濾并在真空干燥箱中干燥,所得殘余物過硅膠柱純化,得到化合物中間體ii;所述中間體i與的摩爾比為1:1.0~1.5,pd(oac)2與中間體i的摩爾比為0.001~0.02:1,k3po4與中間體i的摩爾比為1.0~2.0:1,dmf用量為:中間體i:dfm=1g:10~20ml;第三步反應方程式第三步:氮氣氛圍下,稱取中間體ii溶解于n,n-二甲基甲酰胺中,再將及醋酸鈀加入,攪拌混合物,再加入磷酸鉀水溶液,將上述反應物的混合溶液于反應溫度120-150℃下加熱回流10-24小時;反應結束后,冷卻加水、將混合物過濾并在真空干燥箱中干燥,所得殘余物過硅膠柱純化,得到目標化合物;所述中間體ii與的摩爾比為1:1.0~1.5,pd(oac)2與中間體ii的摩爾比為0.001~0.02:1,k3po4與中間體ii的摩爾比為1.0~2.0:1,dmf用量為:中間體ii:dfm=1g:15~30ml。本申請人還提供了一種所述的有機化合物用于制備有機電致發光器件的應用方法。本申請人還提供了一種有機電致發光器件,所述有機電致發光器件包括至少一層功能層含有所述的以三嗪和苯并咪唑為核心的有機化合物。本申請人還提供了一種有機電致發光器件,包括空穴阻擋層/電子傳輸層,所述空穴阻擋層/電子傳輸層含有所述的以三嗪和苯并咪唑為核心的有機化合物本申請人還提供了一種有機電致發光器件,包括cpl層即光提取層,所述cpl層含有所述的以三嗪和苯并咪唑為核心的有機化合物。本申請人還提供了一種照明或顯示元件,包括所述的有機電致發光器件。本發明有益的技術效果在于:本發明的有機化合物的結構含有三嗪和苯并咪唑兩種剛性基團,提升了材料結構穩定性;本發明材料的分子量在700-850之間,在空間結構上,三嗪4和6位為強電子性的苯并咪唑基團,2位以空穴基團分開,使得材料具有較高的密度,獲得了較高的折射率;在可見光領域吸收低、折射率高;同時,使得本發明材料都具有很高的tg溫度和分子熱穩定性;700-850分子量的材料在真空狀態下的蒸鍍溫度一般都小于350℃,既保證了材料在量產時長時間蒸鍍材料不分解,又降低了由于蒸鍍溫度的熱輻射對蒸鍍mask的形變影響。本發明材料在oled器件中應用在cpl層,不參與器件的電子和空穴傳輸,但對材料的熱穩定性、膜結晶性及光傳輸(高折射率)具有非常高的要求。如上分析,三嗪和苯并咪唑為剛性基團,提高了材料的穩定性;高的tg溫度,保證了材料在薄膜狀態下不結晶;低的蒸鍍溫度,是材料可應用于量產的前提;高的折射率則是本發明材料能應用于cpl層的最主要因素。本發明材料由于具有深的homo能級,高電子遷移率,可有效阻擋空穴或能量從發光層傳遞至電子層一側,從而提高空穴和電子在發光層中的復合效率,從而提升oled器件的發光效率和使用壽命。本發明在應用于oled器件的cpl層后,可有效提升oled器件的光取出效率。綜上,本發明所述化合物在oled發光器件中具有良好的應用效果和產業化前景。附圖說明圖1為本發明所列舉的材料應用于oled器件的結構示意圖;其中,1、oled器件基板,2、陽極層,3、空穴注入層,4、空穴傳輸層,5、發光層,6、空穴阻擋層/電子傳輸層,7、電子注入層,8、陰極層,9、cpl層。圖2為化合物76的折射率測試圖;圖3為化合物32和公知材料cbp的膜加速實驗對比圖。具體實施方式實施例1:中間體i的合成氮氣氛圍下,稱取原料2,4,6-三氯-1,3,5-三嗪溶解于四氫呋喃中,再將ar1的硼酸化合物及四(三苯基膦)鈀加入,攪拌混合物,再加入飽和碳酸鉀水溶液,將上述反應物的混合溶液于反應溫度70-90℃下加熱回流10-20小時;反應結束后,冷卻、混合液用二氯甲烷萃取,萃取物用無水硫酸鈉干燥處理,且在減壓下濃縮,將濃縮固體過硅膠柱純化,得到化合物中間體i;所述2,4,6-三氯-1,3,5-三嗪與ar1-b(oh)2的摩爾比為1:1.0~1.5,pd(pph3)4與2,4,6-三氯-1,3,5-三嗪的摩爾比為0.005~0.05:1,k2co3與2,4,6-三氯-1,3,5-三嗪的摩爾比為1.0~2.0:1,thf用量為2,4,6-三氯-1,3,5-三嗪:thf=1g:10~20ml;以中間體a1合成為例:在250ml三口瓶中,通入氮氣,加入0.04mol原料2,4,6-三氯-1,3,5-三嗪,100ml的thf,0.05mol2-萘硼酸,0.0004mol四(三苯基膦)鈀,攪拌,然后加入0.06mol的k2co3水溶液(2m),加熱至80℃,回流反應15小時,取樣點板,反應完全。自然冷卻,用200ml二氯甲烷萃取,分層,萃取液用無水硫酸鈉干燥,過濾,濾液旋蒸,過硅膠柱純化,得到中間體a1,hplc純度99.5%,收率82.8%。元素分析結構(分子式c13h7cl2n3):理論值c,56.55;h,2.56;cl,25.68;n,15.22;測試值:c,56.56;h,2.58;cl,25.65;n,15.21。esi-ms(m/z)(m+):理論值為275.00,實測值為275.27。以中間體a1的合成方法制備中間體i,具體結構如表1所示。表1實施例2:中間體的合成當ar2或ar3表示為通式(2)結構時,(1)在250ml三口瓶中,通入氮氣,加入0.02mol原料2-溴-苯并咪唑、0.03mol碘苯、0.04mol氫化鈉、0.004mol碘化亞銅和0.01mol鄰菲羅啉溶解在100ml1,3-二甲基-2-咪唑啉酮中,攪拌反應20-30h,反應結束后,加水并用二氯甲烷萃取,有機層用無水硫酸鈉干燥,用石油醚和乙酸乙酯的混合物為淋洗劑淋洗,淋洗劑中石油醚與乙酸乙酯的體積比為1:100,柱層析純化,得中間體m;(2)氮氣氛圍下,稱取中間體m溶解于四氫呋喃中,再將br-ar2-b(oh)2及四(三苯基膦)鈀加入,攪拌混合物,再加入飽和碳酸鉀水溶液,將上述反應物的混合溶液于反應溫度70-90℃下加熱回流10-20小時;反應結束后,冷卻、混合液用二氯甲烷萃取,萃取物用無水硫酸鈉干燥處理,且在減壓下濃縮,將濃縮固體過硅膠柱純化,得到化合物中間體n;(3)氮氣氛圍下,稱取中間體n溶解于n,n-二甲基甲酰胺(dmf)中,再將雙(頻哪醇根基)二硼、(1,1’-雙(二苯基膦)二茂鐵)二氯鈀(ii)以及乙酸鉀加入,攪拌混合物,將上述反應物的混合溶液于反應溫度120-150℃下加熱回流5-10小時;反應結束后,冷卻、且將混合物過濾并在真空烘箱中干燥。將所獲得的殘余物過硅膠柱分離純化,得到化合物中間體iv;當ar2或ar3表示為通式(3)結構時,(1)氮氣氛圍下,稱取2-溴-苯并咪唑溶解于四氫呋喃中,再將ar5-b(oh)2及四(三苯基膦)鈀加入,攪拌混合物,再加入飽和碳酸鉀水溶液,將上述反應物的混合溶液于反應溫度70-90℃下加熱回流5-15小時;反應結束后,冷卻、混合液用二氯甲烷萃取,萃取物用無水硫酸鈉干燥處理,且在減壓下濃縮,將濃縮固體過硅膠柱純化,得到化合物中間體p;(2)氮氣氛圍下,加入中間體p、i-ar2-br、氫化鈉、碘化亞銅和鄰菲羅啉溶解在1,3-二甲基-2-咪唑啉酮中,攪拌反應20-30h,反應結束后,加水并用二氯甲烷萃取,有機層用無水硫酸鈉干燥,用石油醚和乙酸乙酯的混合物為淋洗劑淋洗,柱層析純化,得中間體q;(3)氮氣氛圍下,稱取中間體q溶解于n,n-二甲基甲酰胺(dmf)中,再將雙(頻哪醇根基)二硼、(1,1’-雙(二苯基膦)二茂鐵)二氯鈀(ii)以及乙酸鉀加入,攪拌混合物,將上述反應物的混合溶液于反應溫度120-150℃下加熱回流5-10小時;反應結束后,冷卻、且將混合物過濾并在真空烘箱中干燥。將所獲得的殘余物過硅膠柱分離純化,得到化合物中間體iv;當ar2或ar3表示為通式(4)結構時,(1)氮氣氛圍下,加入原料w、i-ar7、氫化鈉、碘化亞銅和鄰菲羅啉溶解在1,3-二甲基-2-咪唑啉酮中,攪拌反應20-30h,反應結束后,加水并用二氯甲烷萃取,有機層用無水硫酸鈉干燥,用石油醚和乙酸乙酯的混合物為淋洗劑淋洗,柱層析純化,得中間體x;(2)氮氣氛圍下,稱取中間體x溶解于四氫呋喃中,再將ar6-b(oh)2及四(三苯基膦)鈀加入,攪拌混合物,再加入飽和碳酸鉀水溶液,將上述反應物的混合溶液于反應溫度70-90℃下加熱回流5-15小時;反應結束后,冷卻、混合液用二氯甲烷萃取,萃取物用無水硫酸鈉干燥處理,且在減壓下濃縮,將濃縮固體過硅膠柱純化,得到化合物中間體y;(3)氮氣氛圍下,稱取中間體y溶解于n,n-二甲基甲酰胺中,再將br-ar2-b(oh)2及醋酸鈀加入,攪拌混合物,再加入磷酸鉀水溶液,將上述反應物的混合溶液于反應溫度120-150℃下加熱回流10-24小時;反應結束后,冷卻加水、將混合物過濾并在真空干燥箱中干燥,所得殘余物過硅膠柱純化,得化合物化合物中間體z;(4)氮氣氛圍下,稱取中間體z溶解于n,n-二甲基甲酰胺(dmf)中,再將雙(頻哪醇根基)二硼、(1,1’-雙(二苯基膦)二茂鐵)二氯鈀(ii)以及乙酸鉀加入,攪拌混合物,將上述反應物的混合溶液于反應溫度120-150℃下加熱回流5-10小時;反應結束后,冷卻、且將混合物過濾并在真空烘箱中干燥。將所獲得的殘余物過硅膠柱分離純化,得到化合物中間體iv;以中間體b7合成為例(1)在250ml三口瓶中,通入氮氣,加入0.02mol原料2-溴-5-氯-1h-苯并咪唑、0.03mol碘苯、0.04mol氫化鈉、0.004mol碘化亞銅和0.01mol鄰菲羅啉溶解在100ml1,3-二甲基-2-咪唑啉酮中,攪拌反應20-30h,反應結束后,加水并用二氯甲烷萃取,有機層用無水硫酸鈉干燥,用石油醚和乙酸乙酯的混合物為淋洗劑淋洗,淋洗劑中石油醚與乙酸乙酯的體積比為1:100,柱層析純化,得中間體x1;hplc純度99.7%,收率78.5%。元素分析結構(分子式c13h8brcln2):理論值c,50.76;h,2.62;br,25.98;cl,11.53;n,9.11;測試值:c,50.74;h,2.63;br,25.96;cl,11.55;n,9.12。esi-ms(m/z)(m+):理論值為305.96,實測值為306.24。(2)在250ml三口瓶中,通入氮氣,加入0.04mol中間體x1,100ml的thf,0.05mol苯硼酸,0.0004mol四(三苯基膦)鈀,攪拌,然后加入0.06molk2co3水溶液(2m),加熱至80℃,回流反應10小時,取樣點板,反應完全。自然冷卻,用200ml二氯甲烷萃取,分層,萃取液用無水硫酸鈉干燥,過濾,濾液旋蒸,過硅膠柱純化,得到中間體y1,hplc純度99.8%,收率88.2%。元素分析結構(分子式c19h13cln2):理論值c,74.88;h,4.30;cl,11.63;n,9.19;測試值:c,74.84;h,4.33;cl,11.65;n,9.18。esi-ms(m/z)(m+):理論值為304.08,實測值為304.52。(3)在250ml三口瓶中,通入氮氣,加入0.02mol中間體y1,120ml的dmf,0.04mol苯硼酸,0.0002mol醋酸鈀,攪拌,然后加入0.02molk3po4水溶液,加熱至130℃,回流反應10小時,取樣點板,反應完全。自然冷卻,用200ml二氯甲烷萃取,分層,萃取液用無水硫酸鈉干燥,過濾,濾液旋蒸,過硅膠柱純化,得到中間體z1,hplc純度99.5%,收率80.5%。元素分析結構(分子式c25h17brn2):理論值c,70.60;h,4.03;br,18.79;n,6.59;測試值:c,70.60;h,4.05;br,18.78;n,6.57。esi-ms(m/z)(m+):理論值為424.06,實測值為424.34。(4)在500ml三口瓶中,通入氮氣,加入0.05mol中間體z溶解于300ml的n,n-二甲基甲酰胺(dmf)中,再將0.06mol雙(頻哪醇根基)二硼、0.0005mol(1,1’-雙(二苯基膦)二茂鐵)二氯鈀(ii)以及0.125mol乙酸鉀加入,攪拌混合物,將上述反應物的混合溶液于反應溫度120-150℃下加熱回流10小時;反應結束后,冷卻并加入200ml水、且將混合物過濾并在真空烘箱中干燥。將所獲得的殘余物過硅膠柱分離純化,得到化合物中間體b7;hplc純度99.2%,收率81.2%。元素分析結構(分子式c31h29bn2o2):理論值c,78.82;h,6.19;b,2.29;n,5.93;o,6.77;測試值:c,78.92;h,6.14;b,2.25;n,5.94;o,6.75。esi-ms(m/z)(m+):理論值為472.23,實測值為472.59。以中間體b7的合成方法制備中間體iv,具體結構如表2所示。表2實施例3:化合物1的合成:在250ml三口瓶中,通入氮氣,加入0.01mol中間體a1,150ml的dmf,0.03mol中間體b1,0.0002mol醋酸鈀,攪拌,然后加入0.02molk3po4水溶液,加熱至150℃,回流反應24小時,取樣點板,反應完全。自然冷卻,用200ml二氯甲烷萃取,分層,萃取液用無水硫酸鈉干燥,過濾,濾液旋蒸,過硅膠柱純化,得到目標產物,hplc純度99.1%,收率72.1%。元素分析結構(分子式c51h33n7):理論值c,82.35;h,4.47;n,13.18;測試值:c,82.37;h,4.48;n,13.15。esi-ms(m/z)(m+):理論值為743.28,實測值為743.62。實施例4:化合物10的合成:在250ml三口瓶中,通入氮氣,加入0.01mol中間體a2,150ml的dmf,0.03mol中間體b2,0.0002mol醋酸鈀,攪拌,然后加入0.02molk3po4水溶液,加熱至150℃,回流反應24小時,取樣點板,反應完全。自然冷卻,用200ml二氯甲烷萃取,分層,萃取液用無水硫酸鈉干燥,過濾,濾液旋蒸,過硅膠柱純化,得到目標產物,hplc純度99.3%,收率75.4%。元素分析結構(分子式c55h47n7):理論值c,81.96;h,5.88;n,12.16;測試值:c,81.99;h,5.86;n,12.15。esi-ms(m/z)(m+):理論值為805.39,實測值為805.74。實施例5:化合物13的合成:化合物13的制備方法同實施例3,不同之處在于用中間體a3替換中間體a1。元素分析結構(分子式c53h35n7):理論值c,82.68;h,4.58;n,12.74;測試值:c,82.68;h,4.56;n,12.76。esi-ms(m/z)(m+):理論值為769.30,實測值為769.67。實施例6:化合物19的合成:化合物19的制備方法同實施例3,不同之處在于用中間體a7替換中間體a1;中間體b8代替中間體b1。元素分析結構(分子式c45h29n9):理論值c,77.68;h,4.20;n,18.12;測試值:c,77.68;h,4.20;n,18.12。esi-ms(m/z)(m+):理論值為695.25,實測值為695.25。實施例7:化合物25的合成:化合物25的制備方法同實施例3,不同之處在于用中間體a5替換中間體a1。元素分析結構(分子式c56h39n7):理論值c,83.04;h,4.85;n,12.11;測試值:c,83.01;h,4.86;n,12.13。esi-ms(m/z)(m+):理論值為809.33,實測值為809.66。實施例8:化合物27的合成:化合物27的制備方法同實施例3,不同之處在于用中間體a6替換中間體a1。元素分析結構(分子式c53h34n8):理論值c,81.31;h,4.38;n,14.31;測試值:c,81.31;h,4.36;n,14.33。esi-ms(m/z)(m+):理論值為782.29,實測值為782.67。實施例9:化合物31的合成:化合物31的制備方法同實施例3,不同之處在于用中間體a7替換中間體a1。元素分析結構(分子式c47h31n7):理論值c,81.36;h,4.50;n,14.13;測試值:c,81.33;h,4.52;n,14.15。esi-ms(m/z)(m+):理論值為693.26,實測值為693.58。實施例10:化合物32的合成:化合物32的制備方法同實施例9,不同之處在于用中間體b3替換中間體b1。元素分析結構(分子式c47h31n7):理論值c,81.36;h,4.50;n,14.13;測試值:c,81.35;h,4.51;n,14.14。esi-ms(m/z)(m+):理論值為693.26,實測值為693.67。實施例11:化合物39的合成:化合物39的制備方法同實施例9,不同之處在于用中間體b4替換中間體b1。元素分析結構(分子式c53h43n7):理論值c,81.83;h,5.57;n,12.60;測試值:c,81.83;h,5.54;n,12.63。esi-ms(m/z)(m+):理論值為777.36,實測值為777.67。實施例12:化合物48的合成:化合物48的制備方法同實施例10,不同之處在于用中間體a8替換中間體a7。元素分析結構(分子式c53h35n7):理論值c,82.68;h,4.58;n,12.74;測試值:c,82.67;h,4.56;n,12.77。esi-ms(m/z)(m+):理論值為769.30,實測值為769.73。實施例13:化合物53的合成:在250ml三口瓶中,通入氮氣,加入0.01mol中間體a7,150ml的dmf,0.015mol中間體b1,0.0001mol醋酸鈀,攪拌,然后加入0.01molk3po4水溶液,加熱至150℃,回流反應24小時,取樣點板,反應完全。自然冷卻,用200ml二氯甲烷萃取,分層,萃取液用無水硫酸鈉干燥,過濾,濾液旋蒸,過硅膠柱純化,得到中間體c1,hplc純度99.2%,收率85.1%。元素分析結構(分子式c28h18cln5):理論值c,73.12;h,3.94;cl,7.71;n,15.23;測試值:c,73.13;h,3.96;cl,7.70;n,15.21。esi-ms(m/z)(m+):理論值為459.13,實測值為459.37。在250ml三口瓶中,通入氮氣,加入0.01mol中間體c1,150ml的dmf,0.015mol中間體b5,0.0001mol醋酸鈀,攪拌,然后加入0.01molk3po4水溶液,加熱至150℃,回流反應24小時,取樣點板,反應完全。自然冷卻,用200ml二氯甲烷萃取,分層,萃取液用無水硫酸鈉干燥,過濾,濾液旋蒸,過硅膠柱純化,得到目標產物,hplc純度99.5%,收率71.7%。元素分析結構(分子式c47h31n7):理論值c,81.36;h,4.50;n,14.13;測試值:c,81.34;h,4.51;n,14.15。esi-ms(m/z)(m+):理論值為693.26,實測值為693.71。實施例14:化合物59的合成:化合物59的制備方法同實施例10,不同之處在于用中間體a9替換中間體a7。元素分析結構(分子式c59h38n8):理論值c,82.50;h,4.46;n,13.04;測試值:c,82.51;h,4.47;n,13.02。esi-ms(m/z)(m+):理論值為858.32,實測值為858.72。實施例15:化合物63的合成:化合物63的制備方法同實施例9,不同之處在于用中間體b5替換中間體b1。元素分析結構(分子式c47h31n7):理論值c,81.36;h,4.50;n,14.13;測試值:c,81.33;h,4.53;n,14.14。esi-ms(m/z)(m+):理論值為693.26,實測值為693.65。實施例16:化合物70的合成:化合物70的制備方法同實施例13,不同之處在于用中間體b3替換中間體b5。元素分析結構(分子式c47h31n7):理論值c,81.36;h,4.50;n,14.13;測試值:c,81.32;h,4.54;n,14.14。esi-ms(m/z)(m+):理論值為693.26,實測值為693.66。實施例17:化合物76的合成:化合物76的制備方法同實施例9,不同之處在于用中間體b6替換中間體b1。元素分析結構(分子式c47h31n7):理論值c,81.36;h,4.50;n,14.13;測試值:c,81.37;h,4.52;n,14.11。esi-ms(m/z)(m+):理論值為693.26,實測值為693.54。實施例18:化合物83的合成:化合物83的制備方法同實施例15,不同之處在于用中間體a10替換中間體a7。元素分析結構(分子式c51h39n7):理論值c,81.68;h,5.24;n,13.07;測試值:c,81.71;h,5.25;n,13.04。esi-ms(m/z)(m+):理論值為749.33,實測值為749.64。實施例19:化合物92的合成:化合物92的制備方法同實施例18,不同之處在于用中間體a11替換中間體a10。元素分析結構(分子式c59h38n8):理論值c,82.50;h,4.46;n,13.04;測試值:c,82.51;h,4.47;n,13.02。esi-ms(m/z)(m+):理論值為858.32,實測值為858.73。實施例20:化合物94的合成:化合物94的制備方法同實施例13,不同之處在于用中間體b7替換中間體b5。元素分析結構(分子式c53h35n7):理論值c,82.68;h,4.58;n,12.74;測試值:c,82.70;h,4.57;n,12.73。esi-ms(m/z)(m+):理論值為769.30,實測值為769.62。本發明的有機化合物在發光器件中使用作為cpl層材料,具有高的tg(玻璃轉化溫度)溫度和高折射率。對本發明化合物及現有材料分別進行熱性能及折射率測試,結果如表3所示。其中化合物76的折射率測試圖如圖2所示。表3注:玻璃化溫度tg由示差掃描量熱法(dsc,德國耐馳公司dsc204f1示差掃描量熱儀)測定,升溫速率10℃/min;折射率是由橢偏儀(美國j.a.woollamco.型號:alpha-se)測量,測試為大氣環境。由上表數據可知,對比目前應用的cbp、alq3及tpbi等材料,本發明的有機化合物具有高的玻璃轉化溫度、高折射率,同時由于含有三嗪和苯并咪唑剛性基團,保證了材料的熱穩定性。因此,本發明以三嗪和苯并咪唑為核心的有機材料在應用于oled器件的cpl層后,可有效提高器件的光取出效率,并且保證了oled器件的長壽命。以下通過器件實施例1~21和器件比較例1詳細說明本發明合成的oled材料在器件中的應用效果。本發明所述器件實施例2~21、器件比較例1與器件實施例1相比所述器件的制作工藝完全相同,并且所采用了相同的基板材料和電極材料,電極材料的膜厚也保持一致,所不同的是器件實施例2~18對器件中的cpl層材料做了變換;器件實施例19~21對器件的空穴阻擋/電子傳輸層材料做了變換,各實施例所得器件的性能測試結果如表4所示。器件實施例1:一種電致發光器件,其制備步驟如圖1所示包括:a)清洗透明oled器件基板1上的ito陽極層2,分別用去離子水、丙酮、乙醇超聲清洗各15分鐘,然后在等離子體清洗器中處理2分鐘;b)在ito陽極層2上,通過真空蒸鍍方式蒸鍍空穴注入層材料hat-cn,厚度為10nm,這層作為空穴注入層3;c)在空穴注入層3上,通過真空蒸鍍方式蒸鍍空穴傳輸材料npb,厚度為80nm,該層為空穴傳輸層4;d)在空穴傳輸層4之上蒸鍍發光層5,cbp作為作為主體材料,ir(ppy)3作為摻雜材料,ir(ppy)3和cbp的質量比為1:9,厚度為30nm;e)在發光層5之上,通過真空蒸鍍方式蒸鍍電子傳輸材料tpbi,厚度為40nm,這層有機材料作為空穴阻擋/電子傳輸層6使用;f)在空穴阻擋/電子傳輸層6之上,真空蒸鍍電子注入層lif,厚度為1nm,該層為電子注入層7;g)在電子注入層7之上,真空蒸鍍陰極mg:ag/ag層,mg:ag摻雜比例為9:1,厚度15nm,ag厚度3nm,該層為陰極層8;h)在陰極層8之上,通過真空蒸鍍方式蒸鍍cpl材料化合物1,厚度為50nm,這層有機材料作為cpl層9使用。按照上述步驟完成電致發光器件的制作后,測量器件的電流效率和壽命,其結果見表4所示。相關材料的分子機構式如下所示:器件實施例2:電致發光器件的cpl層材料變為本發明化合物10。器件實施例3:電致發光器件的cpl層材料變為本發明化合物13。器件實施例4:電致發光器件的cpl層材料變為本發明化合物19。器件實施例5:電致發光器件的cpl層材料變為本發明化合物25。器件實施例6:電致發光器件的cpl層材料變為本發明化合物27。器件實施例7:電致發光器件的cpl層材料變為本發明化合物31。器件實施例8:電致發光器件的cpl層材料變為本發明化合物32。器件實施例9:電致發光器件的cpl層材料變為本發明化合物39。器件實施例10:電致發光器件的cpl層材料變為本發明化合物48。器件實施例11:電致發光器件的cpl層材料變為本發明化合物53。器件實施例12:電致發光器件的cpl層材料變為本發明化合物59。器件實施例13:電致發光器件的cpl層材料變為本發明化合物63。器件實施例14:電致發光器件的cpl層材料變為本發明化合物70。器件實施例15:電致發光器件的cpl層材料變為本發明化合物76。器件實施例16:電致發光器件的cpl層材料變為本發明化合物83。器件實施例17:電致發光器件的cpl層材料變為本發明化合物92。器件實施例18:電致發光器件的cpl層材料變為本發明化合物94。器件實施例19:電致發光器件的空穴阻擋/電子傳輸層材料變為本發明化合物19。器件實施例20:電致發光器件的空穴阻擋/電子傳輸層材料變為本發明化合物52。器件實施例21:電致發光器件的空穴阻擋/電子傳輸層材料變為本發明化合物85。器件比較例1:電致發光器件的cpl層材料變為公知材料alq3。所得電致發光器件的檢測數據見表4所示。表4由表4的結果可以看出本發明所述以三嗪和苯并咪唑為核心的有機化合物應用于oled發光器件制作后,與器件比較例1相比,光取出得到明顯提升,相同電流密度下,器件亮度和器件效率都得到了提升,由于亮度及效率得到提升,oled器件在定亮度下的功耗相對降低,使用壽命也得到提高。為了說明本發明材料膜相態結晶穩定性能,將本發明材料化合物32和公知材料cbp進行了膜加速結晶實驗:采用真空蒸鍍方式,分別蒸鍍將化合物32和cbp蒸鍍在無堿玻璃上,并在手套箱(水氧含量<0.1ppm)中進行封裝,將封裝后樣品在雙85(溫度85℃,濕度85%)條件下進行放置,定期用顯微鏡(leica,dm8000m,5*10倍率)觀察材料膜的結晶狀態,實驗結果如表5所示,材料表面形態如圖3所示:表5材料名稱化合物32cbp材料成膜后表面形態光滑平整均勻表面形態光滑平整均勻實驗72小時后表面形態光滑平整均勻,無結晶表面形成若干分散的圓形結晶面實驗600小時后表面形態光滑平整均勻,無結晶表面龜裂以上實驗說明,本發明材料的膜結晶穩定性遠遠高于公知材料,在應用于oled器件后的使用壽命具有有益效果。當前第1頁12
相關技術
網友詢問留言
已有0條留言
- 還沒有人留言評論。精彩留言會獲得點贊!
1