一種水溶性單核鎳配合物與制備方法及其應用與流程
本發(fā)明屬于鎳配合物領域,尤其是涉及一種水溶性單核鎳配合物與制備方法及其應用。
背景技術:
自上世紀90年代以來,藥物小分子與生物大分子dna、蛋白質相互作用的研究己經成為眾多科研人員的共同關注和感興趣的課題,因為進行藥物小分子(包括潛在藥物)與dna、蛋白質之間相互作用研究不僅有助于人們從分子水平上認識和理解藥物與生物大分子相互作用的方式及其在體內作用機制,而且對抗癌藥物的體外篩選具有重要意義,同時也為設計療效更好、毒性更低的抗癌新藥提供理論指導和有價值的信息。
癌癥是嚴重威脅人類健康甚至生命的一類疾病,積極預防、診治是廣大科研學者的共同目標。而小分子與dna的相互作用是以dna為靶分子的,其鍵合狀態(tài)一方面可能導致癌變、突變及細胞死亡,比如一些致癌物能與dna形成加合物,這種dna加合物是可能癌變的預警標志物,另一方面也可能是臨床上廣泛應用的抗癌藥物,例如腫瘤組織是細胞分裂活躍的部位,而許多抗癌藥物都是以dna為作用靶點,通過與癌細胞的dna發(fā)生相互作用破壞其結構,進而影響基因調控與表達功能,表現出抗癌活性。此外,小分子化合物與dna特異性的定位結合,在基因表達的調控過程及很多抗癌藥物體內作用方式的研究中非常重要。因此小分子與dna的相互作用的研究不僅有利于探索和開發(fā)新的核酸探針,而且有助于從分子水平上了解抗癌藥物的作用機理,闡明有毒物的致癌、致畸的分子生物學機理,為設計臨床上更為有效地抗癌藥物提供理論指導。
利用n-甲基-n,n-二吡啶甲基胺(bpma)為配體,以金屬鎳離子為活性中心,通過加熱回流攪拌的合成方法,設計合成了一個新的水溶性單核鎳配合物;并使用元素分析、紅外光譜、x-單晶衍射分析、紫外-可見光譜、熒光光譜等手段對配合物進行表征和性質研究,發(fā)現配合物能夠以插入方式與小牛胸腺dna發(fā)生相互作用,并且能夠和bsa發(fā)生較強地相互作用。
技術實現要素:
有鑒于此,本發(fā)明旨在提出一種水溶性單核鎳配合物與制備方法及其應用,采用加熱回流攪拌在室溫條件下,采用緩慢揮發(fā)的合成方法得到目標產物,制備方法簡單可靠。
為達到上述目的,本發(fā)明的技術方案是這樣實現的:
一種水溶性單核鎳配合物,化學式為[ni(bpma)2](clo4)2,其中bpma為n-甲基-n,n-二吡啶甲基胺。
進一步的,所述該配合物的結構式為:
進一步的,所述該配合物屬于正交晶系,p212121空間點群,晶胞參數為
進一步的,所述該配合物為單核結構,ni1金屬離子分別與來自兩個bpma配體的氮原子n1、n2、n3、n4、n5、n6配位,形成一個畸變的八面體構型,其中n2、n3、n4、n5組成八面體的赤道平面,n1和n6占據八面體的軸向位置;ni1-n1、ni1-n2、ni1-n3、ni1-n4、ni1-n5、ni1-n6的鍵長分別為
本發(fā)明還提供了一種制備如上所述的水溶性單核鎳配合物的方法,包括如下步驟:
將nicl2·6h2o溶解于乙腈和水組成的混合溶液中,在上述溶液中加入含有bpma的乙腈溶液,然后加熱回流攪拌,冷卻至室溫后;滴加naclo4的飽和水溶液,繼續(xù)攪拌,然后過濾;濾液置于室溫條件下,緩慢揮發(fā)數周后,得到適于x-單晶衍射分析的紫色塊狀晶體,經乙醚洗滌后、干燥。
進一步的,所述nicl2·6h2o與bpma的物質的量比為(0.2~0.5mmol):(0.4~1.0mmol)。
進一步的,所述加熱回流攪拌的反應時間為2~6小時,溫度為82~100℃。
進一步的,所述乙腈和水組成的混合溶液中乙腈與水的體積比為1.0:1.0~2.0。
進一步的,所述含有bpma的乙腈溶液的滴加速度為0.05ml/s。
本發(fā)明同時提供了一種如上所述的水溶性單核鎳配合物或如上所述的制備方法制備的水溶性單核鎳配合物在制備核酸識別試劑和潛在藥物中的應用。
產物元素分析的實驗值分別為:c45.63%、h4.40%、n12.30%。其中元素分析的理論值分別為:c45.60%、h4.38%、n12.28%。
用kbr壓片的紅外光譜分析,該配合物在1607cm-1處出現很尖的強吸收峰,為bpma上的c–n伸縮振動產生的吸收;在2964cm-1和2901cm-1的兩個中等強度的尖銳吸收峰可以指派為bpma的c-h伸縮振動;622~760cm-1多重峰為吡啶環(huán)的伸縮振動峰;1081cm-1的強峰和622cm-1的中強峰可認為高氯酸根沒有參與配位,與單晶結構相符。
通過配合物與ct-dna相互作用的吸收光譜研究和熒光淬滅實驗證實所述單核鎳配合物以部分插入的中等鍵合方式和dna發(fā)生相互作用。
通過配合物與bsa相互作用的吸收光譜研究證實所述單核鎳配合物能夠與bsa發(fā)生較強地相互作用。
相對于現有技術,本發(fā)明所述的水溶性單核鎳配合物與制備方法及其應用具有以下優(yōu)勢:
本發(fā)明所述的水溶性單核鎳配合物與制備方法及其應用,制備步驟簡單,加熱回流攪拌、室溫緩慢揮發(fā)條件下即可得到配合物的單晶,配合物具有較好的水溶性,從而可以在生理條件下研究其生物活性,發(fā)現配合物能夠以部分插入的鍵合方式和dna發(fā)生相互作用,可以作為潛在的核酸識別試劑,并且發(fā)現配合物還能夠與bsa發(fā)生明顯的相互作用,可以能夠作為潛在的藥物。
附圖說明
圖1為本發(fā)明實施例1所述的水溶性單核鎳配合物的晶體結構圖;
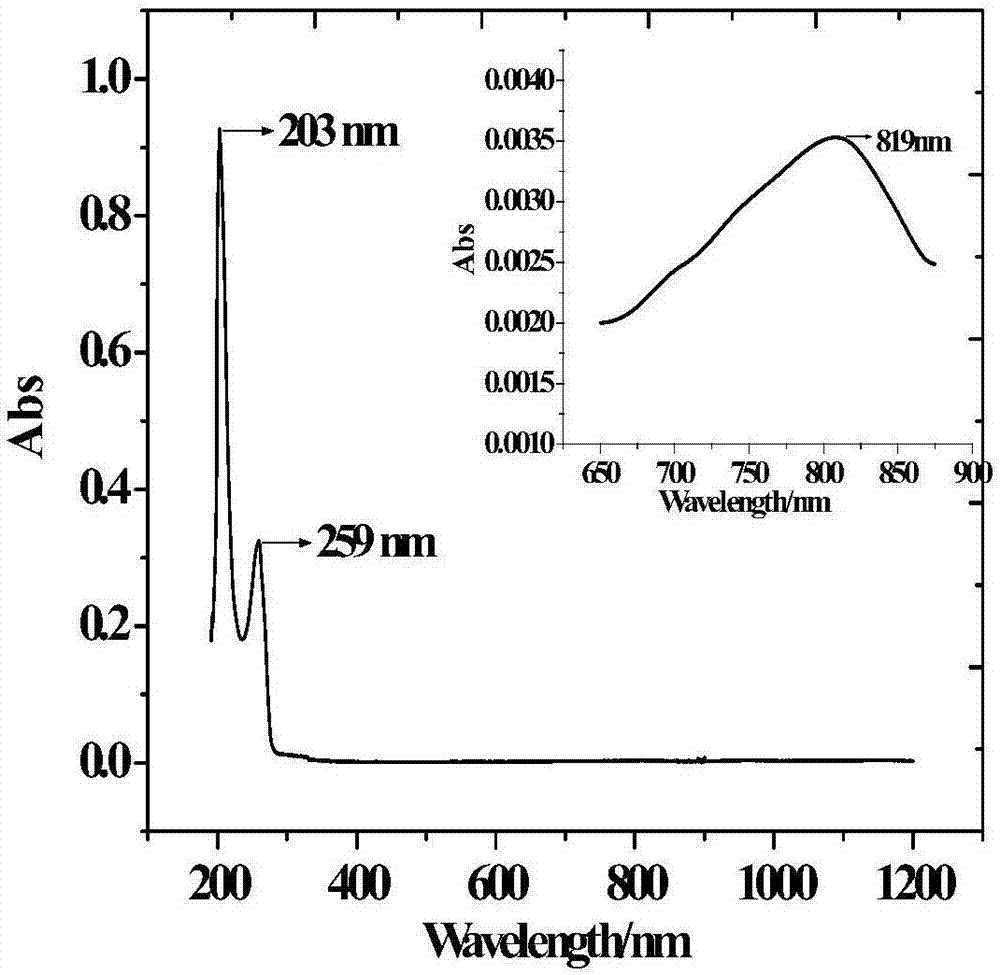
圖2為本發(fā)明實施例1所述的水溶性單核鎳配合物的紫外可見光譜圖;
圖3為本發(fā)明實施例1所述的水溶性單核鎳配合物在加入不同量的ct-dna后的電子吸收光譜圖;
圖4為在273k和298k下,加入不同濃度的本發(fā)明實施例1所述的水溶性單核鎳配合物的eb-dna的熒光淬滅光譜圖;
圖5為本發(fā)明實施例1所述的水溶性單核鎳配合物在273k和298k對eb-dna復合物熒光的影響;
圖6為本發(fā)明實施例1所述的水溶性單核鎳配合物加入不同量的bsa后的電子吸收光譜圖。
具體實施方式
除有定義外,以下實施例中所用的技術術語具有與本發(fā)明所屬領域技術人員普遍理解的相同含義。以下實施例中所用的試驗試劑,如無特殊說明,均為常規(guī)生化試劑;所述實驗方法,如無特殊說明,均為常規(guī)方法。
下面結合實施例及附圖來詳細說明本發(fā)明。
實施例1
水溶性單核鎳配合物的合成:
將0.3mmolnicl2·6h2o溶解于15ml由乙腈和水按照體積比1:1組成的混合溶液中,然后以0.05ml/s的滴速逐滴向上述溶液中加入1.00ml其中含有0.6mmolbpma的乙腈溶液,隨后加熱回流攪拌3小時,待冷卻至室溫后,滴加2.50mlnaclo4的飽和水溶液,室溫下繼續(xù)攪拌半小時后過濾,濾液置于室溫條件下,緩慢揮發(fā)后,得到適于x-單晶衍射的紫色塊狀晶體,用乙醚洗滌后干燥,產率為:49%。
水溶性單核鎳配合物的測試:
1、產物元素分析的實驗值分別為:c45.57%、h4.46%、n12.24%。其中元素分析的理論值分別為:c45.60%、h4.38%、n12.28%,元素分析結果與晶體結構式相符合。
2、紫外-可見光譜分析:
如圖2所示,從配合物的紫外可見光譜圖中可以看出,配合物在200~1200nm的范圍內有三個吸收帶,分別是:203nm處的吸收,為π-π*躍遷,ε=6.5×103l·mol-1·cm-1,屬于較強吸收;259nm處的吸收,為配體到金屬離子的n-π荷移躍遷,ε=2.2×103l·mol-1·cm-1,屬于較強吸收;819nm處的吸收,為金屬離子的d-d躍遷,ε=27.1l·mol-1·cm-1,屬于弱吸收。
3、ft-ir(kbr):用kbr壓片的紅外光譜分析,配合物在2964cm-1和2901cm-1的出現的中等強度、寬峰指派為bpma的c-h伸縮振動峰;在1607cm-1處出現尖、強峰,為bpma上的c–n伸縮振動峰;622~760cm-1多重峰為吡啶環(huán)的伸縮振動峰;1081cm-1的強峰和622cm-1的中強峰推測出高氯酸根沒有參與配位,與單晶結構相符。
4、鎳配合物的晶體結構測定:
1)實驗過程:
在296(2)k下,選取大小合適的晶體,用經石墨單色器單色化的mo-kα
2)實驗結果:
如圖1所示,該配合物為單核結構,x-射線單晶衍射分析數據顯示,在配合物中,金屬離子ni1的配位數為六,分別和來自兩個bpma配體的氮原子n1,n2,n3,n4,n5,n6配位,形成畸變八面體構型。其中n2,n3,n4,n5組成八面體的赤道平面,n1和n6占據在八面體的軸向位置,ni1-n1、ni1-n2、ni1-n3、ni1-n4、ni1-n5、ni1-n6的鍵長分別是
表1配合物的晶體結構的主要數據
表2配合物晶體的主要鍵長、鍵角
5、實施加入不同量的ct-dna,觀察水溶性單核鎳配合物的電子吸收光譜變化試驗:
1)實驗過程:
在室溫下,向樣品池和參比池中各加2.0ml緩沖溶液,緩沖溶液用三蒸水配制其中含有50mm的nacl和5mm的tris,然后用鹽酸調至ph=7.4,然后向樣品池加入一定量體積的配合物溶液,配合物濃度為1.45×10-4m,并向參比池中補加相應等體積的緩沖溶液。用微量進樣器向樣品池和參比池中加入一定量相同體積的ct-dna儲備液,使ct-dna與配合物的濃度比值不斷增加,ct-dna的濃度為0~3.94×10-4m,觀察配合物吸收峰的變化。
2)實驗結果:
如圖3所示,配合物在204nm處有強的紫外吸收,隨著ct-dna的逐漸等量加入,配合物的最大紫外吸收強度出現明顯的下降,即出現了明顯的“減色效應”,同時伴隨了紅移現象,紅移距離△λ為5nm,減色率為39.2%,說明配合物在本實驗條件下,以部分插入的鍵合方式和dna發(fā)生相互作用。
6、實施加入不同量的鎳配合物,觀察eb-dna的熒光淬滅試驗:
鎳配合物本身不產生熒光,采用配合物淬滅eb-dna結合物的熒光,通過研究eb-dna結合物熒光強度的變化來間接測定配合物與dna的結合程度。
1)實驗過程:
配制ct-dna和eb的混合溶液,其含量分別為2.4×10-6meb和4.8×10-5mct-dna,儲備于4℃冰箱中,鎳配合物配制成3.0×10-3m儲備液。淬滅滴定實驗時,向樣品池中加入2.0ml儲備的eb-dna混合液,觀察其熒光強度,然后用微量進樣器每次向樣品池中加入等體積的配合物儲備液,觀察發(fā)射光譜的變化并將數據保存以便擬合處理。
2)實驗結果:
如圖4所示,單核鎳配合物淬滅eb-dna的熒光光譜,在加入鎳配合物后,eb-dna的熒光強度出現明顯的降低,并且隨著配合物濃度的增加,其熒光強度逐漸降低,表明鎳配合物與ct-dna的競爭結合取代了eb。
根據stern-volmer方程式:i0/i=1+k[q],以加入單核鎳配合物前后eb-dna的熒光強度比值(i0/i)為縱坐標,鎳配合物的濃度為橫坐標,做出了stern-volmer圖即圖5,配合物的熒光淬滅曲線呈直線,基本上滿足了stern-volmer方程,表明熒光淬滅是鎳配合物取代了已插入dna的eb的結果。根據方程keb[eb]=kapp[complex],keb=1.0×107m-1([eb]=4.0μm),計算出鎳配合物的表觀鍵合常數分別為1.40×105m-1。結果顯示配合物與dna是中等鍵合模式,小于溴化乙錠、典型的插入試劑的鍵合常數107m-1。
更進一步分析了在298k和273k下,鎳配合物對eb-dna復合物熒光的影響。如圖5所示,鎳配合物隨著溫度升高,斜率k增大,說明該鎳配合物與eb-dna形成的復合物隨著溫度的升高越來越穩(wěn)定,熒光淬滅是由于擴散作用產生的,其熒光淬滅為動態(tài)淬滅機理。
7、實施加入不同量的bsa,觀察鎳配合物的電子吸收光譜變化試驗:
紫外-可見吸收光譜法主要是利用血清白蛋白中色氨酸、苯丙氨酸、絡氨酸等氨基酸殘基吸收峰的變化來進行分析的,如果吸收峰在小分子配合物和血清白蛋白作用的前后發(fā)生了變化,則說明小分子配合物和血清白蛋白發(fā)生了作用。由此,我們通過電子吸收光譜研究了鎳配合物與bsa的相互作用。
1)實驗過程:
在室溫下,向樣品池和參比池中各加2.0ml緩沖溶液,緩沖溶液用三蒸水配制的濃度為10mmnah2po4/na2hpo4,ph=7.4,然后向樣品池加入一定量體積的鎳配合物溶液并向參比池中補加相應等體積的緩沖溶液。用微量進樣器向樣品池和參比池中加入一定量相同體積的bsa儲備液,使bsa與鎳配合物的濃度比值不斷增加,觀察鎳配合物吸收峰的變化并將數據保存。
2)實驗結果:
如圖6所示,可以清晰地看出,隨著bsa的不斷加入,鎳配合物在207nm處的π-π躍遷峰發(fā)生明顯的減色現象并伴有紅移,紅移的距離為5nm;259nm處的荷移躍遷峰幾乎未受bsa濃度增加的影響。說明在本實驗條件下,表明該鎳配合物與bsa之間發(fā)生了較強地相互作用。
實施例2
實驗步驟:
將0.1mmolnicl2·6h2o溶解于15ml由乙腈和水按照體積比2:3組成的混合溶液中,然后以0.05ml/s的滴速逐滴向上述溶液中加入0.50ml其中含有0.2mmolbpma的乙腈溶液,隨后加熱回流攪拌5小時,待冷卻至室溫后,滴加2.50mlnaclo4的飽和水溶液,室溫下繼續(xù)攪拌半小時,過濾,濾液置于室溫條件下,緩慢揮發(fā)。
實驗結論:
一個月后得到淺紫色的微晶,不適于x-單晶衍射分析,未得到精細結構數據,用乙醚洗滌后干燥,產率為:27%。
實施例3
實驗步驟:
將0.6mmolnicl2·6h2o溶解于15ml由乙腈和水按照體積比1:2組成的混合溶液中,然后以0.05ml/s的滴速逐滴向上述溶液中加入2.00ml其中含有1.2mmolbpma的乙腈溶液,加熱回流攪拌4小時,待冷卻至室溫后,滴加2.50mlnaclo4的飽和水溶液,室溫下繼續(xù)攪拌半小時,過濾,濾液置于4℃冰箱中,緩慢揮發(fā)。
實驗結論:
五周后得到淺紫色的固體粉末,未得到適合結構解析的單晶,用乙醚洗滌后干燥,產率為:51%。
以上所述僅為本發(fā)明的較佳實施例而已,并不用以限制本發(fā)明,凡在本發(fā)明的精神和原則之內,所作的任何修改、等同替換、改進等,均應包含在本發(fā)明的保護范圍之內。
- 還沒有人留言評論。精彩留言會獲得點贊!