雙層倍半硅氧烷環氧樹脂改性劑及其制備方法和應用與流程
本發明涉及環氧樹脂應用領域,具體涉及一種雙層倍半硅氧烷環氧樹脂改性劑及其制備方法和應用。
背景技術:
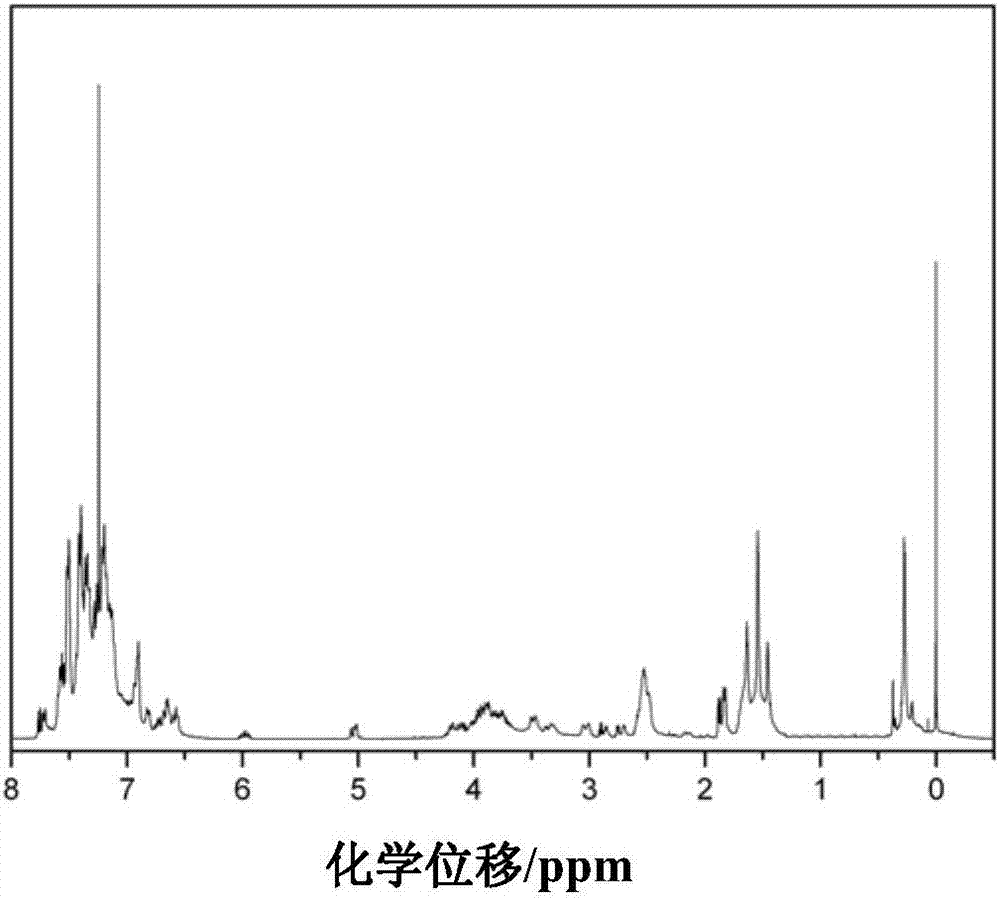
:環氧樹脂具有優異的機械性能、電氣性能和粘結性能而被廣泛應用于結構復合材料、電子半導體封裝、膠粘劑等領域。但是環氧樹脂由于內應力大,本體比較脆,耐濕熱性能較差,斷裂韌性和沖擊強度欠缺,很大程度上限制了其應用。因此對環氧樹脂的增韌改性和耐濕熱改性對環氧樹脂的應用具有很重要的意義。針對環氧樹脂的增韌問題,很多相關方法被開發出來。研究表明采用長鏈熱塑性樹脂、納米粒子和有機硅化合物等進行增韌改性,效果明顯。如公開號為cn104725782a的中國專利公開了一種納米橡膠環氧樹脂增韌劑的制備方法。該方法通過納米橡膠粒子和其他助劑在預分散、細分散、浸潤包覆和超聲波處理等工藝后得到了一種粘度適中、易于分散的增韌改性劑。專利號為cn105906814a的中國專利采用了一種端環氧基團的聚醚作為環氧樹脂的增韌劑。但是由于長鏈聚醚的存在,環氧樹脂的玻璃化溫度急劇降低,對其應用可能產生一定的影響。有機硅具有熱穩定性好、耐候性、耐高低溫性能良好、分子柔順性好等優點,在環氧樹脂中引入有機硅能很好的增韌環氧樹脂并對其綜合性能有所改善。如公開號為cn1560106a的中國專利公開了一種有機硅改性環氧樹脂的制備方法,有效提高了環氧樹脂的韌性和耐水性。但是由于該方法直接采用氯硅烷進行制備增韌劑,可能殘余有機氯可能會對環氧封裝膠的電氣性能有一定的影響。且通常有機硅的玻璃化溫度很低,引入有機硅可能會大大影響環氧樹脂的玻璃化溫度,從而對環氧樹脂的應用范圍產生影響。倍半硅氧烷是一種真正分子水平上的有機無機納米雜化材料,其分子結構由si-o-si形成的主鏈和有機基團形成的側鏈組成。目前,已有人報道采用倍半硅氧烷對環氧樹脂進行改性,尤其以籠型倍半硅氧烷(poss)的研究較多。如公開號為cn102492116a的中國專利文獻中公開了一種環氧樹脂和多面體籠型倍半硅氧烷納米雜化材料,該雜化材料是將γ-(2,3-環氧丙氧)丙基三甲氧基八籠型倍半硅氧烷作為納米粒子添加到環氧樹脂中,在固化劑作用下,八環氧基poss和環氧樹脂發生交聯反應,從而將八環氧基poss單體引入到環氧樹脂結構中,形成了網絡交聯結構。制備得到的雜化材料表現出極低的介電常數和介電損耗值。但由于八環氧基poss的剛性結構,在提高雜化材料的耐熱性能、機械性能的同時,往往會降低雜化材料的韌性。技術實現要素:本發明公開了一種雙層倍半硅氧烷環氧樹脂改性劑,將其應用于環氧樹脂中,在不影響環氧樹脂的玻璃化轉變溫度的同時,可以提高其韌性及耐濕熱性能。具體技術方案如下:一種雙層倍半硅氧烷環氧樹脂改性劑,結構式如下式(ⅰ)或(ⅱ)所示:上式中,n選自1~20的整數,r選自相較于其它類型的倍半硅氧烷,本發明制備的雙層倍半硅氧烷環氧樹脂改性劑的環氧值為0.2~0.3mol/100g,其分子結構中具有多個環氧基團,可以有效的分散并反應到環氧樹脂體系中;另一方面,結構中的雙層倍半硅氧烷結構呈柔性特征,可以有效提高改性環氧樹脂的韌性。本發明公開了上述的雙層倍半硅氧烷環氧樹脂改性劑的制備方法。當所述雙層倍半硅氧烷環氧樹脂改性劑的結構式如式(ⅰ)所示,制備方法包括如下步驟:以含氫雙層倍半硅氧烷和烯丙基雙酚a環氧樹脂為原料,在催化劑作用下進行硅氫加成反應,再經后處理得到;當所述雙層倍半硅氧烷環氧樹脂改性劑的結構式如式(ⅱ)所示,制備方法包括如下步驟:以含氫雙層倍半硅氧烷和烯丙基雙酚a環氧樹脂為原料,在催化劑作用下進行硅氫加成反應,向硅氫加成反應后的產物中加入含雙鍵環氧化合物進行封端反應,再經后處理得到。作為優選,所述含氫雙層倍半硅氧烷和烯丙基雙酚a環氧樹脂的質量比為1:0.5~2;所述的催化劑選自含鉑催化劑,如氯鉑酸、卡斯特催化劑、二氧化鉑等;所述硅氫加成反應的溫度為70~90℃,時間為12~24h。作為優選,所述的含雙鍵環氧化合物選自烯丙基縮水甘油醚、丁香酚基縮水甘油醚等。作為優選,所述的后處理包括減壓蒸餾。作為優選,所述含氫雙層倍半硅氧烷的制備方法如下:a、將苯基三甲氧基硅烷、氫氧化鈉、水混合,回流反應后,經過濾干燥得到中間產物;所述苯基三甲氧基硅烷、氫氧化鈉和水的摩爾比為4:2:5;b、將步驟a得到的中間產物和三乙胺溶解在四氫呋喃溶劑中,降溫至0℃以下,通氮氣并加入甲基二氯硅烷,冰水浴反應后,再經后處理得到所述的含氫雙層倍半硅氧烷;所述中間產物、三乙胺和甲基二氯硅烷的摩爾比為1:1:2。作為優選,所述烯丙基雙酚a環氧樹脂的制備工藝如下:將一定比例的環氧氯丙烷、烯丙基雙酚a和芐基三乙基氯化銨混合后升溫至60~80℃。干燥氮氣保護下將氫氧化鈉水溶液緩慢加入,而后反應1~5h,升溫至100~120℃后將體系中的水分除去,而后降溫至50~70℃,持續反應10~15h。將得到的產物用蒸餾水洗滌至中性,并蒸餾出過量的環氧氯丙烷,即得到所述的烯丙基雙酚a環氧樹脂。所述的環氧氯丙烷、烯丙基雙酚a和芐基三乙基氯化銨的摩爾比為1:0.05~0.1:0.02~0.03。本發明公開了所述的雙層倍半硅氧烷環氧樹脂改性劑的應用,步驟如下:將環氧樹脂、固化劑和所述的雙層倍半硅氧烷環氧樹脂改性劑分散在有機溶劑中,加熱除去有機溶劑后,再進行固化處理。作為優選,所述的雙層倍半硅氧烷環氧樹脂改性劑,其n值選自1或2。經試驗發現,當n值選自上述范圍時,該改性劑可以有效分散在環氧樹脂中,并且對環氧樹脂的耐水性能、力學性能和韌性等都有明顯改善。作為優選,所述的環氧樹脂選自雙酚a型環氧樹脂、雙酚f型環氧樹脂、雙酚s型環氧樹脂中的至少一種;所述的固化劑選自4,4'-二氨基二苯基甲烷和/或3,3'-二氨基二苯基甲烷。與現有技術相比,本發明具有如下優點:本發明合成了一種新型的雙層倍半硅氧烷環氧樹脂改性劑,其分子結構中具有多個環氧基團,可以有效的分散并反應到環氧樹脂體系中;另一方面,結構中的雙層倍半硅氧烷結構呈柔性特征,可以有效提高改性環氧樹脂的韌性;因此可以作為改性劑應用于環氧樹脂的改性領域,相較于其它類型的倍半硅氧烷,它具有分散性好、分子柔順性好的特點,在不影響環氧樹脂的玻璃化轉變溫度的同時,可以提高其韌性及耐濕熱性能。附圖說明圖1為實施例1制備的雙層倍半硅氧烷環氧樹脂改性劑的核磁共振硅譜;圖2為實施例1制備的雙層倍半硅氧烷環氧樹脂改性劑的核磁共振氫譜;圖3為實施例3制備的雙層倍半硅氧烷環氧樹脂改性劑的核磁共振硅譜;圖4為實施例3制備的雙層倍半硅氧烷環氧樹脂改性劑的核磁共振氫譜。具體實施方式下面結合具體實施案例對本發明進行進一步描述,但本發明的保護范圍并不僅限于此:以下實施例中,含氫雙層倍半硅氧烷和烯丙基雙酚a環氧樹脂采用相同的制備工藝,具體如下:含氫雙層倍半硅氧烷的制備工藝是將苯基三甲氧基硅烷、氫氧化鈉、水按照一定比例混合,以異丙醇為溶劑回流反應4h后過濾并干燥得到ddsq-na;其中苯基三甲氧基硅烷、氫氧化鈉、水摩爾比為4:2:5。將ddsq-na和三乙胺在四氫呋喃為溶劑下冰水浴降溫至0℃以下,通氮氣并緩慢加入甲基二氯硅烷,冰水浴反應1h,經過后處理即可制得含氫雙層倍半硅氧烷;其中ddsq-na、三乙胺和甲基二氯硅烷摩爾比為1:1:2;后處理工藝為依次進行水洗、鹽酸洗、飽和碳酸氫鈉洗,水洗三遍,蒸餾后再用100ml甲醇洗三遍并真空干燥即得到產物。烯丙基雙酚a環氧樹脂制備工藝是將摩爾比為1:0.05:0.02的環氧氯丙烷、烯丙基雙酚a和芐基三乙基氯化銨混合后升溫至70℃。干燥氮氣保護下將20%氫氧化鈉水溶液緩慢加入,而后反應2h,升溫至110℃采用分水器將體系中的水分除去,而后降溫至60℃,持續反應10h以上。將得到的產物用蒸餾水洗滌至中性,并蒸餾出過量的環氧氯丙烷,即得到烯丙基雙酚a環氧樹脂。實施例1將10g烯丙基雙酚a環氧樹脂和0.14g卡斯特催化劑(50ppm)加入到100ml甲苯中,氮氣保護下升溫至75℃,而后將溶解于100ml甲苯中的14.4g含氫雙層倍半硅氧烷在2h內緩慢加入,滴加完成后繼續反應12h。減壓蒸餾除去溶劑即可得到雙層倍半硅氧烷環氧樹脂改性劑,記為epddsq-1,n值為1(式i型)。本實施例制備產物的硅譜及氫譜分別如圖1、2所示,并通過滴定環氧值來確定其分子結構中的環氧值為2.01mmol/g。將50ge51環氧樹脂,18.5gdds環氧樹脂固化劑和3.45gepddsq-1在50℃下溶解于丙酮中分散均勻,而后在90℃下真空除去溶劑,將其澆鑄于鋼制模具中,在140℃固化2小時,在160℃固化2小時,在180℃固化2小時。脫模的樣品無氣泡,外觀透明無明顯缺陷。樣品的性能列于下表1中。實施例2將10g烯丙基雙酚a環氧樹脂和0.14g卡斯特催化劑(50ppm)加入到100ml甲苯中,氮氣保護下升溫至75℃,而后將溶解于100ml甲苯中的19.2g含氫雙層倍半硅氧烷在2h內緩慢加入,滴加完成后繼續反應12h。減壓蒸餾除去溶劑即可得到產物epddsq2,n值為2(式i型),環氧值為1.68mmol/g。將50ge51環氧樹脂,18.5gdds環氧樹脂固化劑和3.45gepddsq2在50℃下溶解于丙酮中分散均勻,而后在90℃下真空除去溶劑,將其澆鑄于鋼制模具中,在140℃固化2小時,在160℃固化2小時,在180℃固化2小時。脫模的樣品無氣泡,外觀透明無明顯缺陷。樣品的性能列于下表1中。實施例3將10g烯丙基雙酚a環氧樹脂和0.14g卡斯特催化劑(50ppm)加入到100ml甲苯中,氮氣保護下升溫至75℃,而后將溶解于100ml甲苯中的57.6g含氫雙層倍半硅氧烷在2h內緩慢加入,滴加完成后繼續反應12h。而后加入0.63g烯丙基縮水甘油醚并反應6h。減壓蒸餾除去未反應的烯丙基縮水甘油醚和溶劑即可得到產物epddsq3,n值為1(式ii型,式中r基團為),環氧值為1.35mmol/g。本實施例制備產物的硅譜及氫譜分別如圖3和4所示。將50ge51環氧樹脂,18.5gdds環氧樹脂固化劑和3.45gepddsq3在50℃下溶解于丙酮中分散均勻,而后在90℃下真空除去溶劑,將其澆鑄于鋼制模具中,在140℃固化2小時,在160℃固化2小時,在180℃固化2小時。脫模的樣品無氣泡,外觀透明無明顯缺陷。樣品的性能列于下表1中。實施例4將10g烯丙基雙酚a環氧樹脂和0.14g卡斯特催化劑(50ppm)加入到100ml甲苯中,氮氣保護下升溫至75℃,而后將溶解于甲苯中的57.6g含氫雙層倍半硅氧烷在2h內緩慢加入,滴加完成后繼續反應12h。而后加入1.2g丁香酚縮水甘油醚并反應6h。減壓蒸餾除去未反應的丁香酚縮水甘油醚和溶劑即可得到產物epddsq4,n值為1(式ii型,式中r基團為),環氧值為1.26mmol/g。將50ge51環氧樹脂,18.5gdds環氧樹脂固化劑和3.45gepddsq4在50℃下溶解于丙酮中分散均勻,而后90℃下真空除去溶劑,將其澆鑄于鋼制模具中,在140℃固化2小時,在160℃固化2小時,在180℃固化2小時。脫模的樣品無氣泡,外觀透明無明顯缺陷。樣品的性能列于下表1中。對比例將50ge51環氧樹脂,18.5gdds環氧樹脂固化劑在50℃下溶解于丙酮中分散均勻,而后在90℃下真空除去溶劑,將其澆鑄于鋼制模具中,在140℃固化2小時,在160℃固化2小時,在180℃固化2小時。脫模的樣品無氣泡,外觀透明無明顯缺陷。樣品的性能列于下表1中。表1項目實施例1實施例2實施例3實施例4對比例拉伸強度(mpa)89.693.588.389.958.3沖擊強度(kj/m2)25.629.626.326.513.9玻璃化溫度(℃)162163163167169斷裂韌性(mpa·m1/2)1.141.221.091.080.65吸水率(23℃,%)1.10.80.90.82.6吸水率(100℃,%)1.51.21.31.23.5從表1可以看出,加入雙層倍半硅氧烷環氧樹脂改性劑后,與未改性環氧樹脂相比,環氧樹脂的拉伸強度、沖擊強度和斷裂韌性都有顯著提高,吸水率明顯降低。玻璃化轉變溫度沒有明顯下降,顯著改性了環氧樹脂的綜合性能。當前第1頁12
相關技術
網友詢問留言
已有0條留言
- 還沒有人留言評論。精彩留言會獲得點贊!
1