一種4?芳乙醇基?1,8?萘酰亞胺類化合物及其制備方法和應用與流程
本發明涉及到一種4-芳乙醇基-1,8-萘酰亞胺類化合物及其制備方法,屬于含萘酰亞胺熒光團所涉及的有機光電或熒光探針等領域,其中涉及4-芳基乙腈-1,8-萘酰亞胺類化合物可應用在陰離子識別領域,通過紫外光譜且肉眼可見識別氟離子。
背景技術:
陰離子識別,特別是針對氟離子識別,在生命,醫藥,環境等領域有著重要的應用。過量氟離子的攝入可導致牙齒、骨骼組織代謝疾病產生;飲用水中的氟離子超標也是重要的檢測指標;同時,高毒氟離子的檢測在環境及生物體系里有著重要應用。
顏色化學傳感器可通過將分子識別的信息轉換成易被感知的顏色信號,具有靈敏,快速及檢測極限低等特點,可廣泛應用在生物化學、細胞生物學和分析化學等相關領域。
萘酰亞胺類化合物是一類非常優良的發色團,因其具有熒光強烈、色彩鮮艷、熱穩定性好以及高的熒光量子產率等特性,可作為熒光增白劑、熒光染料,在光電敏感材料以及熒光探針等領域有著廣泛的應用。目前報道的萘酰亞胺衍生物主要通過萘酐的酐頭進行酰胺化衍生,以及通過4-溴-1,8-萘酐的溴原子與相應的取代的氨基或羥基等官能團發生一些取代反應進而衍生一些4-位含nh或oh等基團的萘酰亞胺類化合物。
綜上所述,4位含ch基的萘酰亞胺衍生物并未有較多的報道,特別是4-位芳基乙醇基的萘酰亞胺衍生物至今尚未有報道。目前文獻報道的4位合成酰基的萘酰亞胺類衍生物主要采用傅克酰基化反應的方法制得4位芳基ch基衍生物,該傅克酰基化反應方法存在反應條件苛刻且副反應較多,反應收率低等缺點。(ep.2251687a1;polymer45(2004)6445;polymer41(2000)2367;polymerdegradationandstability97(2012)1581;j.org.chem.2010,75,2989;macromolecules2000,33,4310;j.org.chem.1983,48,4097;j.org.chem.2004,69,1364;j.org.chem.2013,78,4974)。
取得4位芳乙醇基-1,8-萘酰亞胺衍生物非常有必要,因為對羥基進行再次衍生可以得到一系列有用的新穎的萘酰亞胺類衍生物。
技術實現要素:
本發明的目的之一是為了解決上述的傅克酰基化反應方法制備4位芳基ch基衍生物過程中存在反應條件苛刻且副反應較多,反應收率低等技術問題而提供一種4-芳乙醇基-1,8-萘酰亞胺類化合物的制備方法,該制備方法具有反應條件溫和、無副反應發生,且反應收率高等優點。
本發明的目的之二在于提供一種上述制備方法所得的4-芳乙醇基-1,8-萘酰亞胺類化合物。
本發明的目的之三在于提供一種上述4-芳酰基-1,8-萘酰亞胺類化合物的合成方法過程中所產生的中間產物4-芳基乙腈-1,8-萘酰亞胺類化合物,及該4-芳基乙腈-1,8-萘酰亞胺類化合物作為檢測陰離子的顏色探針的應用,特別是在作為檢測氟離子的顏色探針的應用。
本發明提供的技術方案
一種4-芳乙醇基-1,8-萘酰亞胺類化合物,其結構通式如下:
其中r1為r1為甲基、乙基、丙基、正丁基、正戊基、正己基、正庚基、正辛基、正壬基或正癸基等烷基;
r2為苯基、萘基、聯苯基、取代苯基、5元或6元的雜芳環基或苯并5元或6元雜芳環基;其中所述5元或6元雜芳基的雜原子為n或/和o,雜原子個數為1或2;
其中,取代苯基為對氟苯基、鄰氯苯基、鄰溴苯基、對甲氧基苯基、間甲氧基苯基或間溴苯基;
5元雜芳基為呋喃基、吡咯基或咪唑基;
6元雜芳基為吡啶基或嘧啶基;
苯并5元雜芳基為苯并呋喃基、苯并咪唑基、boc保護的吲哚基或吲哚基;
苯并6元雜芳基為苯并吡啶基或苯并嘧啶基。
上述4-芳乙醇基-1,8-萘酰亞胺類化合物的合成方法,其合成路線如下所示:
首先,以n-r1-4-溴-1,8萘酰亞胺為起始原料,將n-r1-4-溴-1,8萘酰亞胺與r2取代的乙腈在有機溶劑1中,通過堿催化劑即鈉氫(以下簡稱nah固體)或甲醇鈉催化進行取代反應4-10h,使4位的溴被r2取代的乙腈中的亞甲基取代,得到4-芳基乙腈-1,8-萘酰亞胺類化合物;
然后,將上述所得的4-芳基乙腈-1,8-萘酰亞胺類化合物在有機溶劑2中經堿性陰離子催化劑四丁基氰化胺、四丁基氟化胺或四丁基氫氧化胺作用進行分子內的脫氰基反應得到4-芳酰基-1,8-萘酰亞胺類化合物;
最后,4-芳酰基-1,8-萘酰亞胺類化合物在有機溶劑3中經還原催化劑鈀碳(以下簡稱pd/c)加氫或氯化亞錫作用進行還原反應,最終得到4-芳乙醇基-1,8-萘酰亞胺類化合物。
上述4-芳乙醇基-1,8-萘酰亞胺類化合物的合成方法,具體步驟如下:
(1)、以n-r1-4-溴-1,8萘酰亞胺為起始原料,在有機溶劑1中,在堿催化劑作用下與r2取代的乙腈,室溫、攪拌條件下進行取代反應4-10h,所得的反應液1經硅膠柱層析處理,得到4-芳基乙腈-1,8-萘酰亞胺類化合物;
所述堿催化劑為鈉氫或甲醇鈉;
所述的有機溶劑1為甲苯或四氫呋喃,其用量,按n-r1-4-溴-1,8萘酰亞胺:有機溶劑1為1g:50-60ml的比例計算;
上述n-r1-4-溴-1,8萘酰亞胺與r2取代的乙腈的用量,按n-r1-4-溴-1,8萘酰亞胺:r2取代的乙腈的摩爾比為1:1.1-1.4的比例計算,優選為1:1.114-1.132;
所述的堿催化劑的用量,按n-r1-4-溴-1,8-萘酰亞胺:堿催化劑的摩爾比為1:5.4-6的比例計算,優選為1:5.4;
上述所得的n-r1-4-芳基乙腈-1,8-萘酰亞胺類化合物可用于對陰離子進行檢測,即作為檢測陰離子的顏色探針或熒光探針的應用,特別是作為檢測氟離子或氰根離子的顏色探針或熒光探針;
反應液1經硅膠柱進行層析處理,處理過程用洗脫劑進行洗脫,洗脫劑為石油醚(沸程:60-90℃):乙酸乙酯的體積比為20:1組成的混合液,收集rf=0.4-0.5(石油醚:乙酸乙酯=5:1)階段的出峰,即得4-芳基乙腈-1,8-萘酰亞胺類化合物;
(2)、步驟(1)所得的4-芳基乙腈-1,8-萘酰亞胺類化合物在有機溶劑2中經堿性陰離子催化劑作用,室溫、攪拌條件下進行陰離子誘導分子內脫氰基反應2-10h,所得的反應液2經硅膠柱層析處理,得到4-芳酰基-1,8-萘酰亞胺類化合物;
所述的有機溶劑2為二氯甲烷、乙腈或二甲基甲酰胺等,有機溶劑2的用量,按4-芳基乙腈-1,8-萘酰亞胺類化合物:有機溶劑2為1g:250-300ml的比例計算;
所述的堿性陰離子催化劑為四丁基氰化胺、四丁基氟化胺或四丁基氫氧化胺,優選四丁基氟化銨,其用量,按4-芳基乙腈-1,8-萘酰亞胺類化合物:堿性陰離子催化劑的摩爾比為1:21-25的比例計算,優選為1:21;
反應液2經硅膠柱進行層析處理,處理過程用洗脫劑進行洗脫,洗脫劑為石油醚(沸程:60-90℃):乙酸乙酯的體積比為25:1組成的混合液,收集rf=0.6-0.8(石油醚:乙酸乙酯的體積比為5:1)階段的出峰,即得4-芳酰基-1,8-萘酰亞胺類化合物;
(3)、步驟(2)所得的4-芳酰基-1,8-萘酰亞胺類化合物在有機溶劑3中經還原催化劑作用,室溫、攪拌條件下進行還原反應3-4h,所得的反應液3經硅膠柱層析處理,得到4-芳乙醇基-1,8-萘酰亞胺類化合物;
所述的有機溶劑3為乙腈、甲醇或二甲基甲酰胺等,有機溶劑3的用量,按4-芳酰基-1,8-萘酰亞胺類化合物:有機溶劑3為1g:50-60ml的比例計算;
所述的還原催化劑為pd/c或氯化亞錫,其用量,按4-芳酰基-1,8-萘酰亞胺類化合物:還原催化劑的摩爾比為1:0.1-0.15的比例計算,優選為1:0.1;
上述反應液3經硅膠柱進行層析處理,處理過程用洗脫劑進行洗脫,洗脫劑為石油醚(沸程:60-90℃):乙酸乙酯=25:1組成的混合液,收集rf=0.2-0.3(石油醚:乙酸乙酯=5:1)階段的出峰,即得4-芳乙腈基-1,8-萘酰亞胺類化合物。
由于1,8-萘酰亞胺類化合物在染料及藥物等領域廣泛應用及芳乙醇基可有效地進行官能團的變換,上述所得的4-芳乙醇基-1,8-萘酰亞胺類化合物,可用于合成抗抑郁藥、抗腫瘤藥、抗躁動不安藥物及復雜染料分子的中間體。
本發明的有益效果
本發明的一種4-芳乙醇基-1,8萘酰亞胺類化合物的合成方法,由于以4-溴-1,8萘酰亞胺及取代芳基乙腈為起始原料,該原料屬工業易得原料,因此該合成方法具有生產成本低的特點。
進一步,本發明的一種4-芳乙醇基-1,8萘酰亞胺類化合物的合成方法,由于過程涉及常規的取代反應、氧化反應以及還原反應,因此該合成方法具有合成路線簡單、過程容易控制,最終產物收率高等優點。
進一步,本發明的一種4-芳乙醇基-1,8萘酰亞胺類化合物的合成方法中所得的中間產物4-芳基乙腈-1,8-萘酰亞胺化合物具有識別氟離子的功能,特別是對氟離子具有超高的靈敏性和選擇性。因此可作為氟離子的高選擇的檢測探針,特別地,該類探針能給出4-芳基乙腈-1,8-萘酰亞胺類化合物溶液從無色到藍色的裸眼檢測因此,該4-芳基乙腈-1,8-萘酰亞胺類化合物可作為檢測氟離子的檢測試劑進而取得實際應用。
附圖說明
圖1、應用實施例1中,實施例2所得的4-芳基乙腈-1,8-萘酰亞胺類化合物分別與氟離子,磷酸二氫根離子,醋酸根離子,氯離子,溴離子,碘離子,硫酸氫根離子,硝酸根離子,四氟硼酸根離子,高氯酸根離子作用的乙腈溶液的紫外光譜分析圖;
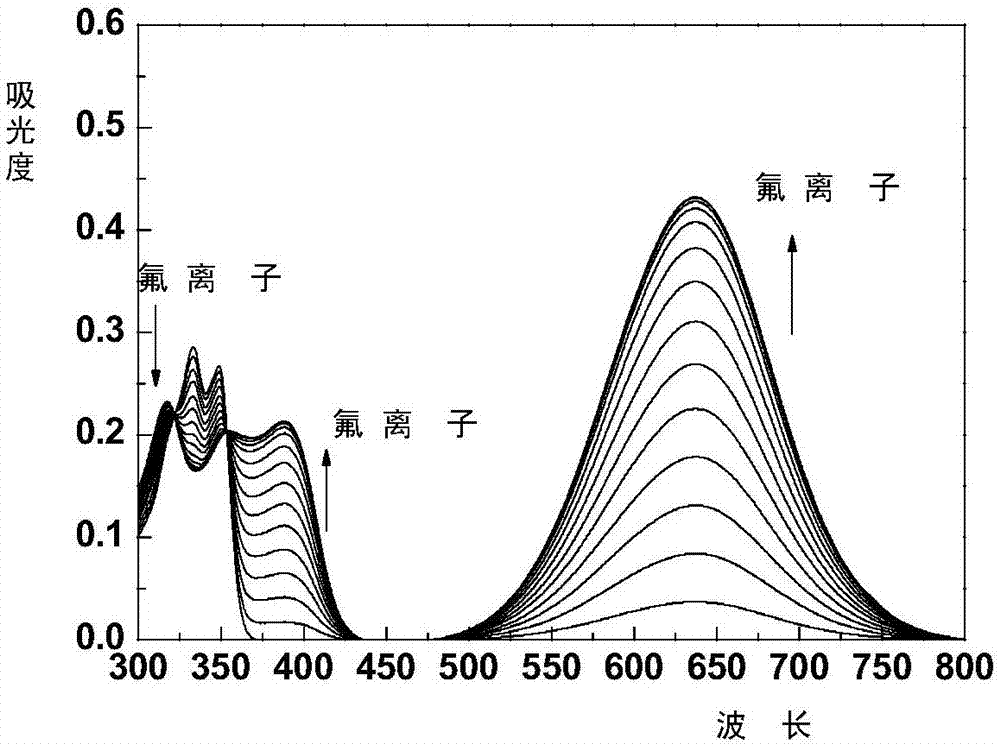
圖2、應用實施例2中,實施例2所得的4-芳基乙腈-1,8-萘酰亞胺類化合物與f-離子連續滴定作用所得的4-芳基乙腈-1,8-萘酰亞胺類化合物的乙腈溶液的紫外滴定圖譜。
具體實施方式
下面通過具體的實施例并結合附圖對本發明進一步闡述,但并不限制本發明。
本發明各實施例中所用的各種原料的名稱、規格及生產廠家的信息請給出(可以以如下表格的形式進行列出)。
本發明的各實施例中所用的硅膠柱的型號及生產廠家為長45cm,直徑45mm,北京聯華玻璃儀器有限公司生產的硅膠柱。
實施例1
一種4-芳乙腈基-1,8-萘酰亞胺類化合物,其結構式如下所示:
其中r1為c4直鏈烷基;
r2為苯基。
上述的一種4-芳乙腈基-1,8-萘酰亞胺類化合物的制備方法,其合成過程的反應方程式如下所示:
上述的一種4-芳基乙腈-1,8-萘酰亞胺類化合物的制備方法,具體步驟如下:
在50ml的三口瓶中,先加入20ml有機溶劑1甲苯及0.40g(3.42mmol)r2取代的乙腈即苯乙腈,0.65g(16.31mmol)堿催化劑nah(60w%),氮氣置換三次,磁力攪拌混勻,然后開始滴加n-正丁基-4-溴-1,8萘酰亞胺(1.0g,3.02mmol)與10ml的有機溶劑1甲苯,滴加用時約30min,滴加完畢,控制溫度為25-30℃、攪拌轉速為500r/min進行取代反應3h,反應過程用tlc點板追蹤至反應完全;
所述的有機溶劑1甲苯的用量,按n-正丁基-4-溴-1,8萘酰亞胺:有機溶劑1為1g:30ml的比例計算;
上述的r2取代的乙腈即苯乙腈的用量,按n-正丁基-4-溴-1,8萘酰亞胺:苯乙腈的摩爾比為1:1.132的比例計算;
所述的堿催化劑nah的用量,按n-正丁基-4-溴-1,8萘酰亞胺:堿催化劑nah的摩爾比為1:5.4的比例計算;
反應完畢后,先用體積百分比濃度為10%的稀鹽酸水溶液調反應液1的ph=1-2,然后用乙酸乙酯萃取,所得的有機相用飽和食鹽水洗滌2次后再用無水硫酸鈉干燥,然后減壓除去溶劑甲苯,然后過硅膠柱進行層析處理,處理過程用洗脫劑進行洗脫,洗脫劑為石油醚(沸程:60-90℃):乙酸乙酯的體積比為20:1組成的混合液,收集rf=0.5(石油醚:乙酸乙酯的體積比為5:1)階段的出峰,最終得0.80g淡黃色固體粉末產品,收率為72.1%。
上述所得的淡黃色固體粉末產品通過核磁共振儀器(brukeravanceiii500mhz)進行氫譜測定,數據如下所示:
1hnmr(500mhz,cdcl3),δ8.56(d,j=6.5,2h),8.18(d,j=8.5hz,1h),7.83(d,j=7.5hz,1h),7.69(t,j=8.0hz,1h),7.30(m,5h),5.83(s,1h),4.11(t,j=7.5hz,2h),1.63(m,2h),1.37(m,2h),0.91(t,j=7.5hz,3h);
上述所得的淡黃色固體粉末產品通過核磁共振儀器(brukeravanceiii500mhz)進行碳譜測定,數據如下所示:
13cnmr(125mhz,cdcl3)δ163.79,163.51,137.56,134.19,131.36,130.80,129.58,129.12,128.94,128.88,127.94,127.84,127.67,123.75,123.69,118.61,100.00,40.38,40.28,30.18,20.36,13.81;
上述所得的淡黃色固體粉末產品通過高分辨質譜儀器(solanx70ft-ms)進行質譜測定,數據如下所示:hrms-esi(m/z):[m+h]+calcd.for(c24h20n2o2),369.16030,found:369.16122.
通過上述所得的淡黃色固體粉末產品的核磁共振譜數據、高分辨質譜方面的數據綜合分析,結果表明,上述所得的淡黃色固體粉末產品為n-正丁基-4-苯乙腈基-1,8-萘酰亞胺,其屬于4-芳基乙腈-1,8-萘酰亞胺類化合物。
實施例2
一種4-芳基乙腈-1,8-萘酰亞胺類化合物,其結構式如下所示:
其中r1為正丁基;
r2為吡啶基。
上述的一種4-芳基乙腈-1,8-萘酰亞胺類化合物的制備方法,其合成過程的反應式如下所示:
上述的一種4-芳基乙腈-1,8-萘酰亞胺類化合物的制備方法,具體步驟如下:
在100ml的三口瓶中,加入25ml有機溶劑1即甲苯、0.50g(3.70mmol)r2取代的乙腈即吡啶-2-乙腈和0.72g(17.92mmol)堿催化劑nah(60w%),氮氣置換三次,室溫攪拌混勻,然后開始滴加n-正丁基-4-溴-1,8萘酰亞胺(1.10g,3.32mmol)與25ml有機溶劑1即甲苯,滴加完畢之后,控制溫度為25-30℃、攪拌轉速為500r/min進行取代反應3h,反應過程用tlc點板追蹤至反應完全;
所述的有機溶劑1甲苯的用量,按n-正丁基-4-溴-1,8萘酰亞胺:有機溶劑1為1g:50ml的比例計算;
上述的r2取代的乙腈即吡啶-2-乙腈的用量,按n-正丁基-4-溴-1,8萘酰亞胺:吡啶-2-乙腈的摩爾比為1:1.114的比例計算;沒問題
所述的堿催化劑nah的用量,按n-正丁基-4-溴-1,8萘酰亞胺:堿催化劑nah的摩爾比為1:5.4的比例計算;
反應完畢后,先用體積百分比濃度為10%的稀鹽酸水溶液調反應液1的ph=1-2,然后用乙酸乙酯萃取,所得的有機相用飽和食鹽水洗滌2次后再用無水硫酸鈉干燥,然后減壓除去溶劑甲苯,然后過硅膠柱層析分離,分離過程洗脫所用的洗脫劑為石油醚(沸程:60-90℃):乙酸乙酯的體積比為25:1組成的混合液,收集rf=0.4(石油醚:乙酸乙酯的體積比為5:1)階段的出峰,最終得0.90g淡黃色固體粉末產品,收率為70.3%.熔點為140.7-140.8℃。
上述所得的淡黃色固體粉末產品通過核磁共振儀器(brukeravanceiii500mhz)進行氫譜測定,數據如下所示:1hnmr(500mhz,cdcl3),δ8.59(d,j=7.5hz,1h),8.54(d,j=6.5hz,2h),8.39(d,j=8.5hz,1h),7.98(d,j=7.5hz,1h),7.70(t,j=7.5hz,1h),7.62(td,j=7.5,1.5hz,1h),7.30(d,j=8.0hz,1h),7.20(dd,j=7.5,5.5hz,2h),6.01(s,1h),4.10(t,j=7.5hz,2h),1.66-1.60(m,2h),1.39-1.33(m,2h),0.90(t,j=7.0hz,1h);
上述所得的淡黃色固體粉末產品通過核磁共振儀器(brukeravanceiii500mhz)進行碳譜測定,數據如下所示:
13cnmr(125mhz,cdcl3)δ163.81,163.50,154.25,150.15,137.80,136.99,131.38,129.97,130.86,129.62,129.01,128.90,127.81,127.52,123.72,123.62,123.58,122.30,118.13,43.43,40.36,30.17,20.35,13.79;
上述所得的淡黃色固體粉末產品通過高分辨質譜儀器(solanx70ft-ms)進行質譜測定,數據如下所示:
hrms-esi(m/z):[m+h]+calcd.for(c23h20n3o2),370.15555,found:370.15634.
通過上述所得的淡黃色固體粉末產品的核磁共振譜數據、高分辨質譜方面的數據綜合分析,結果表明,上述所得的淡黃色固體粉末產品為n-正丁基-4-吡啶乙腈基-1,8-萘酰亞胺,其屬于4-芳基乙腈-1,8-萘酰亞胺類化合物。
上述僅以r2為吡啶基進行舉例,本領域技術人員可以根據上述的實施例,通過調整相應的底物,得到r2為對氟苯基、鄰氯苯基、鄰溴苯基、對甲氧基苯基、間甲氧基苯基或間溴苯基的4-芳基乙腈-1,8-萘酰亞胺類化合物。
實施例3
一種4-芳酰基-1,8-萘酰亞胺類化合物,其結構式如下所示:
其中r1為正丁基;
r2為苯基。
上述的一種4-芳酰基-1,8-萘酰亞胺類化合物的制備方法,其合成過程的反應式如下所示:
上述的一種4-芳酰基-1,8-萘酰亞胺類化合物的制備方法,具體步驟如下:
在50ml的單口瓶中加入實施例1所得的0.04g(0.18mmol)的4-芳基乙腈-1,8-萘酰亞胺類化合物即n-正丁基-4-苯乙腈基-1,8-萘酰亞胺,并加入10ml有機溶劑2即二氯甲烷及1.0g(3.85mmol)堿性陰離子催化劑正四丁基氟化銨,控制溫度為20-25℃,攪拌轉速為500r/min下進行陰離子誘導分子內脫氰基反應4-6h,得到反應液2;
所述的有機溶劑2即二氯甲烷的用量,按4-芳基乙腈-1,8-萘酰亞胺類化合物即n-正丁基-4-苯乙腈基-1,8-萘酰亞胺:有機溶劑2為1g:250ml的比例計算;
所述的堿性陰離子催化劑四丁基氟化胺的用量,按4-芳基乙腈-1,8-萘酰亞胺類化合物即n-正丁基-4-苯乙腈基-1,8-萘酰亞胺:堿性陰離子催化劑的摩爾比為1:21的比例計算;
所得的反應液2減壓蒸去溶劑乙腈,過硅膠柱層析分離,分離過程洗脫采用的洗脫劑為石油醚(沸程:60-90℃):乙酸乙酯的體積比為25:1組成的混合液,收集rf=0.8(石油醚:乙酸乙酯的體積比為5:1)階段的出峰,最終得0.092g淡黃色晶體產品,收率為94.8%。
上述所得的淡黃色晶體產品通過核磁共振儀器(brukeravanceiii500mhz)進行氫譜測定,數據如下所示:
1hnmr(500mhz,cdcl3),δ8.58(d,j=7.5,2h),8.26(d,j=8.0hz,1h),7.78(d,j=7.0hz,2h),7.73(d,j=7.0hz,1h),7.68(t,j=8.0hz,1h),7.59(3,j=7.5hz,1h),7.43(t,j=7.5hz,2h),4.14(t,j=7.5hz,2h),1.67(m,2h),1.39(m,2h),0.92(t,j=7.5hz,3h);
上述所得的淡黃色晶體產品通過核磁共振儀器(brukeravanceiii500mhz)進行碳譜測定,數據如下所示:
13cnmr(125mhz,cdcl3)δ196.32,163.92,163.61,141,88,137.13,134.14,131.80,131.64,130.41,129.73,128.81,128.53,127.95,127.35,124.53,123.01,40.43,30.20,20.38,13.83;
上述所得的淡黃色晶體產品通過高分辨質譜儀器(solanx70ft-ms)進行質譜測定,數據如下所示:
hrms-esi(m/z):[m+h]+calcd.for(c23h19no3),358.14432,found:358.14605.
通過上述所得的淡黃色晶體產品的核磁共振譜數據、高分辨質譜方面的數據綜合分析,結果表明,上述所得的淡黃色晶體產品為n-正丁基-4-苯酰基-1,8-萘酰亞胺,其屬于4-芳酰基-1,8-萘酰亞胺類化合物。
實施例4
一種4-芳酰基-1,8-萘酰亞胺類化合物,其結構式如下所示:
其中r1為正丁基;
r2為吡啶。
上述的一種4-芳酰基-1,8-萘酰亞胺類化合物的制備方法,其合成過程的反應方程式如下所示:
上述的一種4-芳酰基-1,8-萘酰亞胺類化合物的制備方法,具體步驟如下:
50ml的單口瓶中加入實施例2所得的0.04g(0.18mmol)4-芳基乙腈-1,8-萘酰亞胺類化合物即n-正丁基-4-吡啶乙腈基-1,8-萘酰亞胺,并加入10ml有機溶劑2即二氯甲烷及1.0g(3.85mmol)堿性陰離子催化劑正四丁基氟化銨,控制溫度為20-25℃,攪拌轉速為500r/min下進行陰離子誘導分子內脫氰基反應4-6h,得到反應液2;
所述的有機溶劑2即二氯甲烷的用量,按4-芳基乙腈-1,8-萘酰亞胺類化合物即n-正丁基-4-苯乙腈基-1,8-萘酰亞胺:有機溶劑2為1g:250ml的比例計算;
所述的堿性陰離子催化劑四丁基氟化胺的用量,按n-正丁基-4-吡啶乙腈基-1,8-萘酰亞胺:堿性陰離子催化劑四丁基氟化銨的摩爾比為1:21的比例計算;
所得的反應液2減壓蒸去溶劑二氯甲烷,然后過硅膠柱層析分離,分離過程洗脫采用的洗脫劑為石油醚(沸程:60-90℃):乙酸乙酯的體積比為25:1組成的混合液,收集rf=0.6(石油醚:乙酸乙酯的體積比為5:1)階段的出峰,最終得0.09g淡黃色晶體產品,收率為92.7%.熔點為116.6-116.8℃。
上述所得的淡黃色晶體產品通過核磁共振儀器(brukeravanceiii500mhz)進行氫譜測定,數據如下所示:
1hnmr(500mhz,cdcl3),δ8.60-8.56(m,3h),8.31(d,j=8.5hz,1h),8.25(d,j=8.5hz,1h),7.93(td,j=7.5,1.5hz,1h),7.85(d,j=7.5hz,1h),7.68(t,j=7.5hz,1h),7.48(ddd,j=7.0,4.5,1.0hz,1h),4.14(t,j=7.5hz,3h),1.69-1.63(m,2h),1.43-1.35(m,2h),0.92(t,j=7.5hz,3h);
上述所得的淡黃色晶體產品通過核磁共振儀器(brukeravanceiii500mhz)進行碳譜測定,數據如下所示:
13cnmr(125mhz,cdcl3)δ195.66,164.02,165.08,163.84,163.67,154.13,149.34,140.84,137.31,131.86,131.40,129.64,128,04,129.57,128.89,128.50,127.81,127.41,124.71,124.36,122.95,40.37,30.20,20.37,13.83;
上述所得的淡黃色晶體產品通過高分辨質譜儀器(solanx70ft-ms)進行質譜測定,數據如下所示:
hrms-esi(m/z):[m+h]+calcd.for(c23h19fno3),359.13957,found:359.14028.
通過上述所得的淡黃色晶體產品的核磁共振譜數據、高分辨質譜方面的數據綜合分析,結果表明,上述所得的淡黃色晶體產品為n-正丁基-4-吡啶酰基-1,8-萘酰亞胺,其屬于4-芳酰基-1,8-萘酰亞胺類化合物。
上述僅以r2為4-吡啶進行舉例,本領域技術人員可以根據上述的實施例,通過調整相應的底物,得到r2為對氟苯基、鄰氯苯基、鄰溴苯基、對甲氧基苯基、間甲氧基苯基或間溴苯基的4-吡啶酰基-1,8-萘酰亞胺類化合物。
實施例5
一種4-芳乙醇基-1,8-萘酰亞胺類化合物,其結構式如下所示:
其中r1為正丁基;
r2為苯基。
上述的一種4-芳乙醇基-1,8-萘酰亞胺類化合物的制備方法,其合成過程的反應方程式如下所示:
上述的一種4-芳乙醇基-1,8-萘酰亞胺類化合物的制備方法,具體步驟如下:
室溫下向100ml的單口燒瓶內依次加入50ml有機溶劑3即甲醇,實施例3所得的1.00g(2.80mmol)4-芳酰基-1,8-萘酰亞胺類化合物即n-正丁基-4-苯酰基-1,8-萘酰亞胺和0.30g(0.28mmol)還原催化劑pd/c,然后氫氣置換三次,然后控制溫度為25-30℃進行還原反應3-4h,得到反應液3;
所述的有機溶劑3甲醇的用量,按4-芳酰基-1,8-萘酰亞胺類化合物即n-正丁基-4-苯酰基-1,8-萘酰亞胺:有機溶劑3為1g:50ml的比例計算;
所述的還原催化劑為pd/c的用量,按4-芳酰基-1,8-萘酰亞胺類化合物:還原催化劑的摩爾比為1:0.1的比例計算;
所得的反應液3過濾除去催化劑pd/c(10%),然后減壓蒸餾除去甲醇,然后過硅膠柱層析分離,分離過程洗脫采用的洗脫劑為石油醚(沸程:60-90℃):乙酸乙酯的體積比為25:1組成的混合液,收集rf=0.2(石油醚:乙酸乙酯的體積比為5:1)階段的出峰,最終得0.86g淡黃色晶體產品,產率85%。
上述所得的淡黃色晶體產品通過核磁共振儀器(brukeravanceiii500mhz)進行氫譜測定,數據如下所示:
1hnmr(500mhz,cdcl3)δ8.49(d,j=7.5hz,1h),8.44(d,j=7.0hz,1h),8.32(d,j=8.5hz,1h),7.95(d,j=7.6hz,1h),7.95(d,j=7.5hz,1h),7.58(t,j1=8.5hz,1h),7.36-7.27(m,5h),6.53(s,1h),4.10(t,j=8.0hz,2h),3.28(s,1h),1.70-1.64(m,2h),1.46-1.38(m,2h),0.96(t,j=7.0hz,3h).
上述所得的淡黃色晶體產品通過核磁共振儀器(brukeravanceiii500mhz)進行碳譜測定,數據如下所示:
13cnmr(125mhz,cdcl3)δ164.15,163.93,145.99,142.25,130.88,130.73,130.38,128.85,128.76,128.53,128.24,126.97,126.22,125.19,123.07,122.26,73.66,129.11,40.22,30.17,20.34,13.77.
上述所得的淡黃色晶體產品通過高分辨質譜儀器(solanx70ft-ms)進行質譜測定,數據如下所示:
hmrs-esi(m/z):[m+h]+calcd.for(c23h22no3)360.159420;found360.160446.
通過上述所得的淡黃色晶體產品的核磁共振譜數據、紅外光譜及高分辨質譜方面的數據綜合分析,結果表明,上述所得的淡黃色晶體產品為n-正丁基-4-苯乙醇基-1,8-萘酰亞胺,其屬于4-芳乙醇基-1,8-萘酰亞胺類化合物。
實施例6
一種4-芳乙醇基-1,8-萘酰亞胺類化合物,其結構式如下所示:
其中r1為正丁基;
r2為吡啶。
上述的一種4-芳乙醇基-1,8-萘酰亞胺類化合物的制備方法,其合成過程的反應式如下所示:
上述的一種4-芳乙醇基-1,8-萘酰亞胺類化合物的制備方法,具體步驟如下:
室溫下向100ml的單口燒瓶內依次加入50ml有機溶劑3即甲醇,1.00g(2.80mmol)實施例4所得的4-芳酰基-1,8-萘酰亞胺類化合物即n-正丁基-4-吡啶酰基-1,8-萘酰亞胺和0.30g(0.28mmol)還原催化劑pd/c,然后氫氣置換三次,然后控制溫度為25-30℃進行還原反應3-4h,得到反應液3;
所述的有機溶劑3甲醇的用量,按4-芳酰基-1,8-萘酰亞胺類化合物即n-正丁基-4-吡啶酰基-1,8-萘酰亞胺:有機溶劑3為1g:50ml的比例計算;
所述的還原催化劑為pd/c的用量,按4-芳酰基-1,8-萘酰亞胺類化合物即n-正丁基-4-吡啶酰基-1,8-萘酰亞胺:還原催化劑的摩爾比為1:0.1的比例計算;
所得的反應液3過濾除去催化劑pd/c,然后減壓蒸餾除去甲醇,然后過硅膠柱層析分離,分離過程洗脫采用的洗脫劑為石油醚(沸程:60-90℃):乙酸乙酯的體積比為25:1組成的混合液,收集rf=0.3(石油醚:乙酸乙酯的體積比為5:1)階段的出峰,最終得0.99g淡黃色晶體產品,產率98%。
上述所得的淡黃色固體粉末產品通過核磁共振儀器(brukeravanceiii500mhz)進行氫譜測定的數據如下所示:
1hnmr(500mhz,cdcl3)δ8.66(d,j=4.5hz,1h),8.57(dd,j=7.5,2.5hz,2h),8.51(d,j=8.5hz,1h),7.80(d,j=7.5hz,1h),7.68(t,j=8.5hz,1h),7.59(td,j=8.5,1.5hz,1h),7.26(t,j=5.5hz,1h),7.03(d,j=7.5hz,1h),6.45(s,1h),5.57(s,1h),4.17(t,j=7.5hz,2h),1.73-1.67(m,2h),1.48-1.40(m,2h),0.97(t,j=7.0hz,3h).
上述所得的淡黃色固體粉末產品通過核磁共振儀器(brukeravanceiii500mhz)進行碳譜測定的數據如下所示:13cnmr(125mhz,cdcl3)δ164.16,163.92,159.62,148.21,145.25,137.21,130.86,130.85,130.81,129.63,128.89,126.89,126.71,123.21,122.97,122.76,121.25,73.44,40.23,30.18,20.36,13.80;
上述所得的淡黃色固體粉末產品通過高分辨質譜儀器(solanx70ft-ms)進行質譜測定的數據如下所示:
hmrs-esi(m/z):[m+h]+calcd.for(c22h21n2o3)361.154669;found361.154861.
通過上述所得的淡黃色固體粉末產品的核磁共振譜數據、紅外光譜及高分辨質譜方面的數據綜合分析,結果表明,上述所得的淡黃色固體粉末產品為n-正丁基-4-吡啶乙醇基-1,8-萘酰亞胺,其屬于4-芳乙醇基-1,8-萘酰亞胺類化合物。
上述僅以r2為4-吡啶進行舉例,本領域技術人員可以根據上述的實施例,通過調整相應的底物,得到r2為對氟苯基、鄰氯苯基、鄰溴苯基、對甲氧基苯基、間甲氧基苯基或間溴苯基的4-吡啶乙醇基-1,8-萘酰亞胺類化合物。
應用實施例1
稱取18.46mg的實施例2所得的n-正丁基-4-吡啶乙腈基-1,8-萘酰亞胺放入10毫升容量瓶中,用乙腈溶液定容,配置成5mmol/l的溶液。
然后準確量取11份每份體積100ul的上述溶液稀釋到25毫升試管中,然后向上述試管分別一次性加入5.4當量的各種陰離子(從左到右依次為:n-正丁基-4-吡啶乙腈-1,8-萘酰亞胺,n-正丁基-4-吡啶乙腈-1,8-萘酰亞胺+氟離子,n-正丁基-4-吡啶乙腈-1,8-萘酰亞胺+磷酸二氫根離子,n-正丁基-4-吡啶乙腈-1,8-萘酰亞胺+醋酸根離子,n-正丁基-4-吡啶乙腈-1,8-萘酰亞胺+氯離子,n-正丁基-4-吡啶乙腈-1,8-萘酰亞胺+溴離子,n-正丁基-4-吡啶乙腈-1,8-萘酰亞胺+碘離子,n-正丁基-4-吡啶乙腈-1,8-萘酰亞胺+硫酸氫根離子,n-正丁基-4-吡啶乙腈-1,8-萘酰亞胺+硝酸根離子,n-正丁基-4-吡啶乙腈-1,8-萘酰亞胺+四氟硼酸根離子,n-正丁基-4-吡啶乙腈-1,8-萘酰亞胺+高氯酸根離子,加入混合均勻后,迅速移至石英比色皿中,以乙腈作參比溶劑,進行紫外測試,測試結果如圖1所示(圖1中的探針即n-正丁基-4-吡啶乙腈-1,8-萘酰亞胺),從圖1中可以看出,只有加入氟離子的n-正丁基-4-吡啶乙腈-1,8-萘酰亞胺類化合物乙腈溶液有顯著光譜紅移變化,波長移動到了637nm,此外除了醋酸根離子有輕微的紅移現象(吸光度小于0.1且肉眼比色不明顯),其他陰離子的加入無法導致n-正丁基-4-吡啶乙腈-1,8-萘酰亞胺類化合物乙腈溶液的光譜發生變化,且很難通過肉眼檢測到顏色變化,由此表明了n-正丁基-4-吡啶乙腈-1,8-萘酰亞胺類化合物可以通過肉眼高度選擇性識別氟根離子,因此,由此4-芳基乙腈-1,8-萘酰亞胺類化合物可作為高選擇性的氟離子的比色探針。
應用實施例2
將實施例2所得的n-正丁基-4-吡啶乙腈基-1,8-萘酰亞胺配制成20μm的乙腈溶液,同時每次滴加36μl的濃度為5000μm的氟離子乙腈溶液,每次滴加完后分別通過紫外分光光度計記錄所得的紫外吸收譜圖,將多次滴加的圖譜疊加即得到圖2,從圖2中可以看出,隨著氟離子的加入,在637nm及390nm波長處有兩個新的吸收峰并且吸收強度持續增強,而原來在333nm和350nm波長的吸收峰逐漸降低,,這表明n-正丁基-4-吡啶乙腈-1,8-萘酰亞胺類化合物分子與氟離子發生了氫鍵作用,生成了氫鍵復合物,進而影響了探針即n-正丁基-4-吡啶乙腈基-1,8-萘酰亞胺的顏色由無色變成了藍色。
綜上所述,本發明提供的一種4-芳乙醇基-1,8-萘酰亞胺類化合物,如4-苯乙醇基-1,8-萘酰亞胺及4-吡啶乙醇基-1,8-萘酰亞胺,其合成方法簡便,且合成過程中所得的中間產物4-芳基乙腈-1,8-萘酰亞胺化合物,特別是合成過程中的中間產物4-芳基乙腈-1,8-萘酰亞胺化合物可作為氟離子的高選擇的檢測探針,特別地,該類探針能給出4-芳基乙腈-1,8-萘酰亞胺類化合物溶液從無色到藍色的裸眼檢測,因此,該4-芳基乙腈-1,8-萘酰亞胺類化合物可作為檢測氟離子的比色試劑進而取得實際應用。
以上所述內容僅為本發明構思下的基本說明,而依據本發明的技術方案所作的任何等效變換,均應屬于本發明的保護范圍。
- 還沒有人留言評論。精彩留言會獲得點贊!