牛膝糖聚合物及其制備方法和應用與流程
本發明屬于醫藥及保健品技術領域,具體涉及一種牛膝糖聚合物及其制備方法和應用。
背景技術:
骨質疏松癥(osteoporosis,op)是一種以骨量減少,骨微結構受到破壞,同時骨強度降低,從而引起骨的脆性增加和容易于發生骨折為特征的一種疾病。目前防治骨質疏松癥的藥物主要是骨吸收抑制劑、骨形成促進劑和礦化藥物,這些藥物均屬替代治療,在長期使用過程中,存在毒副作用大,靶向性低,依從性差,費用高等缺點。因此,有必要找尋到一種高效、低毒的藥物來治療骨質疏松癥。
牛膝(achyranthesbidentatablume)為莧科植物牛膝的干燥根,被稱為“四大懷藥”之一,具有補肝腎、強筋骨的功效;其主要用于筋骨無力、腰膝酸痛、肝陽眩暈等癥。另外,牛膝還作為活血化瘀的傳統中藥。現代藥理研究表明,其藥理作用較為廣泛,具有抗骨質疏松、抗炎鎮痛、抗腫瘤、促進蛋白質的合成和促進胃腸的興奮性等功效。中醫上有“腎藏精,主骨”的理論,所以一般運用補腎法作為治療骨質疏松癥,已經證實療效顯著。作為滋補腎精類中藥,懷牛膝在治療骨質疏松癥方面的運用已經得到廣泛認可。
高昌琨在現代中藥研究與實踐發表的論文《懷牛膝對維甲酸所致大鼠骨質疏松防治作用的實驗研究》中提到“懷牛膝水煎液能夠在一定程度提高大鼠的活動能力,使得骨中有機質的含量增加,繼而提高骨密度”。同時,牛膝糖聚合物作為牛膝的主要活性成分之一,其結構和活性的報道較少,因此,有必要提供一種提取純化、結構鑒定的方法。
技術實現要素:
為了解決現有技術存在的問題,本發明提供了一種牛膝糖聚合物及其制備方法和應用。本發明制備方法簡單,反應條件溫和,可大規模生產;且本發明對得到的高純度牛膝糖聚合物化學結構進行了鑒定,明確了其成分,為探究其藥理活性機制提供結構依據。同時,本發明得到的牛膝糖聚合物純品為牛膝糖聚合物藥物、質量控制及深入研究其構效關系和作用機制奠定基礎。
本發明提供了一種牛膝糖聚合物,包括牛膝糖聚合物abp3-2,牛膝糖聚合物abw1-1,牛膝糖聚合物abp1-2,牛膝糖聚合物abp4-3。本發明人通過對牛膝糖聚合物粗品提純分離得到牛膝糖聚合物abp3-2,abw1-1,abp1-2,abp4-3,且經結構分析得到其分子量范圍為1000-100000da。
進一步地,所述牛膝糖聚合物abp3-2的結構為:
其中n為1~6或8~30。
進一步地,所述牛膝糖聚合物abw1-1的結構為:
其中x為2~30,y為2~30。
進一步地,所述牛膝糖聚合物abp1-2的結構為:
其中n為1~6或8~30,m為1~11或13~30。
進一步地,所述牛膝糖聚合物abp4-3的結構為:
其中n為1~30,m為1~30,x為1~30,y為1~30。
同時,本發明也提供了一種牛膝糖聚合物的制備方法,包括以下步驟:
s1剪切:將牛膝干燥根切成小段,用水清洗干凈、晾干,得牛膝段;
s2水提:將步驟s1所得牛膝段加入水中,加熱提取,過濾,得提取液和藥渣;
s3分級醇沉:
將步驟s2所得提取液減壓濃縮,得濃縮液1;向濃縮液1中加入乙醇,至乙醇體積濃度為a%,靜置,收集沉淀和上清液,得粗糖聚合物ab1和上清液1;
將上清液1減壓濃縮,得濃縮液2;向濃縮液2中加入乙醇,至乙醇體積濃度為b%,靜置,收集沉淀和上清液,得粗糖聚合物ab2和上清液2;
將上清液2減壓濃縮,得濃縮液3;向濃縮液3中加入乙醇,至乙醇體積濃度為c%,靜置,收集沉淀,得粗糖聚合物ab3;
其中10≤a<b<c<100;
s4堿提:
將步驟s2所得藥渣浸泡于0.1~1m的naoh溶液中,靜置1~10h,收集上清液,得上清液3;
向上清液3中加入0.1~1m的hcl至溶液ph為5~9,離心,收集上清液,減壓濃縮,得濃縮液4;向濃縮液4中加入乙醇,至乙醇體積濃度為50~90%,靜置,收集沉淀,得堿提粗糖聚合物ab4;
s5純化:
一次純化:將步驟s3、s4所得粗糖聚合物ab1、ab2、ab3、ab4進行除蛋白,透析,凍干,得牛膝糖聚合物;
二次純化:
將一次純化后的糖聚合物ab1、ab3、ab4進行離子交換柱層析,用0~2m的nacl進行梯度洗脫,使用苯酚-硫酸法跟蹤洗脫曲線,根據洗脫曲線分別收集糖部分,濃縮、冷凍干燥;再分別用水進行溶解,離心,取上清液;
將上清液再進行分子篩凝膠柱層析,用水進行洗脫,利用苯酚-硫酸法檢測洗脫曲線,根據洗脫曲線收集糖部分,濃縮、冷凍干燥,得abw1-1、abp1-2、abp3-2、abp4-3。
本發明人通過研究發現牛膝段的優選長度為1~3cm;同時,本發明采用水提分級醇沉法和堿提醇沉法的結合,乙醇濃度由低到高進行分級醇沉,對牛膝糖聚合物進行初步分離,且高濃度乙醇能將極性大、水溶性好的糖聚合物與極性小、水溶性差的糖聚合物分離,解決了傳統水煮法提取糖聚合物導致其后期分離繁雜、困難的問題。
進一步地,所述步驟s2中水的添加量為所述牛膝段重量的5~15倍,加熱溫度為60~100℃,提取時間1~10h。
進一步地,所述步驟s3、s4中減壓濃縮溫度為40~70℃,靜置的時間為10~28h,naoh溶液用量為藥渣重量的5~20倍。
另外,本發明還提供了一種牛膝糖聚合物的鑒定方法,包括以下步驟:
(1)取牛膝糖聚合物樣品,完全酸水解,水解產物離子色譜/液相色譜檢測;
(2)取牛膝糖聚合物樣品,干燥,壓片,紅外光譜檢測;
(3)取牛膝糖聚合物樣品,甲基化,水解、還原,乙酰化,進行gc-ms檢測;
(4)取牛膝糖聚合物樣品溶于d2o,進行核磁共振分析。
本發明人為了進一步開發牛膝這一寶貴資源,發掘新藥源,通過大量實驗研究得到本發明:以牛膝干燥根為原料,采用水提醇沉法和堿提醇沉法得到粗糖聚合物,對提取的粗糖聚合物進行脫蛋白,然后利用離子交換層析和凝膠分子篩柱層析方法純化牛膝粗糖聚合物,首次制備出四種牛膝糖聚合物純品,對四種組分糖聚合物純品的理化性質、分子量、單糖組成等進行了系統的分析鑒定,成功得出了四個組分的結構信息,其中牛膝糖聚合物abp3-2由葡萄糖和果糖組成的果聚糖,主鏈由→2)-α-d-fruf-(6→組成,支鏈由→2)-β-d-fruf-(1→組成,末端糖殘基由α-d-glcp-(1→和→2)-β-d-fruf組成;牛膝糖聚合物abw1-1由葡萄糖和果糖組成的果聚糖,主鏈由→2)-α-d-fruf-(1→組成,支鏈由→2)-β-d-fruf-(6→組成,末端糖殘基由α-d-glcp-(1→和→2)-β-d-fruf組成;牛膝糖聚合物abp1-2由葡萄糖和果糖組成的果聚糖,主鏈由→2)-α-d-fruf-(1→組成,支鏈由→2)-β-d-fruf-(6→組成,末端糖殘基由α-d-glcp-(1→和→2)-β-d-fruf組成;牛膝糖聚合物abp4-3由阿拉伯糖,半乳糖,鼠李糖和半乳糖醛酸組成的雜多糖,主鏈由→4)-α-d-galpa-(1→和→2,4)-α-d-galpa-(1→組成,支鏈包括→5)-α-l-araf-(1→、→3)-α-l-araf-(1→、→2,3,5)-α-l-araf-(1→、→6)-β-d-galp-(1→、→3,6)-β-d-galp-(1→、α-l-araf-(1→、α-l-rhap-(1→和β-d-galp-(1→。
本發明還涉及了牛膝糖聚合物在制備治療骨質疏松藥物或保健品中的應用。并通過實驗研究對其抗骨質疏松活性進行評價。
與現有技術相比,本發明具有以下優勢:
1.本發明采用水提醇沉法和堿提醇沉法的結合,對牛膝糖聚合物進行初步分離,效果顯著,且本制備方法簡單,反應條件溫和,可大規模生產;
2.本發明通過柱層析法對牛膝粗糖聚合物進行二次分離純化,效果顯著,首次制備出四種牛膝糖聚合物純品;
3.本發明對純化得到的四種糖聚合物純品的結構進行了鑒定,明確了各糖聚合物組分的理化性質及結構,為探究其藥理活性機制提供結構依據;
4.本發明得到的牛膝糖聚合物純品,組分保存完好,結構明確,質量可控,在斑馬魚骨質疏松模型中顯示具有抗骨質疏松活性,為牛膝糖聚合物在醫藥、保健品等領域的應用提供依據;
5.同時,本發明為牛膝糖聚合物藥物、質量控制及深入研究其構效關系和作用機制奠定基礎。
附圖說明
圖1為abp3-2的離子色譜圖譜圖;
圖2為abp3-2的紅外圖譜;
圖3為abp3-2的1hnmr圖譜;
圖4為abp3-2的13cnmr圖譜;
圖5為abp3-2的hsqc圖譜;
圖6為abp3-2的hmbc圖譜;
圖7為abw1-1的hplc圖譜;
圖8為abw1-1的紅外圖譜;
圖9為abw1-1的1hnmr圖譜;
圖10為abw1-1的13cnmr圖譜;
圖11為abw1-1的hsqc圖譜;
圖12為abw1-1的hmbc圖譜;
圖13為abp1-2的離子色譜圖譜圖譜;
圖14為abp1-2的紅外圖譜;
圖15為abp1-2的1hnmr圖譜;
圖16為abp1-2的13cnmr圖譜;
圖17為abp1-2的hsqc圖譜;
圖18為abp1-2的hmbc圖譜;
圖19為abp4-3的hplc圖譜;
圖20為abp4-3的紅外圖譜;
圖21為abp4-3的1hnmr圖譜;
圖22為abp4-3的13cnmr圖譜;
圖23為abp4-3的hsqc圖譜;
圖24為abp4-3的hmbc圖譜;
圖25為斑馬魚經abw1-1處理后頭部骨骼鈣黃綠素染色的圖像;
圖26為斑馬魚經abw1-1處理后頭部骨骼相對熒光強度的定量分析;
圖27為斑馬魚經abp4-3處理后頭部骨骼鈣黃綠素染色的圖像;
圖28為斑馬魚經abp4-3處理后頭部骨骼相對熒光強度的定量分析。
具體實施方式
下面通過具體實施例對本發明做進一步說明,需要指出的是,以下說明僅僅是對本發明要求保護的技術方案的舉例說明,并非對這些技術方案的任何限制。本發明的保護范圍以所附權利要求書記載的內容為準。
實施例1牛膝糖聚合物的制備方法
所述牛膝糖聚合物的制備方法包括以下步驟:
s1剪切:將10kg的牛膝干燥根用剪刀剪至1~3cm,用冷水快速清洗,晾干,得牛膝段;
s2水提:向步驟s1所得牛膝段中加入其重量10倍的水,加熱至80℃提取,提取2h,收集提取液,藥渣晾干,得提取液和藥渣;
s3分級醇沉:
將步驟s2所得提取液于60℃減壓濃縮后,加入乙醇至乙醇體積濃度為50%,室溫靜置24h后,離心,收集沉淀和上清液,得粗糖聚合物ab1和上清液1;
上清液1于60℃減壓濃縮后,加入乙醇至乙醇體積濃度為70%,室溫靜置24h后,離心,收集沉淀和上清液,得粗糖聚合物ab2和上清液2;
上清液2于60℃減壓濃縮后,加入乙醇至乙醇體積濃度為90%,室溫靜置24h后,離心,收集沉淀和上清液,得粗糖聚合物ab3;
s4堿提:
向步驟s2所得藥渣中加入其重量15倍的0.3m的naoh溶液,室溫放置2h,收集上清液,得上清液3;
向上清液3中加入0.1~1m的hcl至溶液ph為6~8,離心(5000r/min,10min),收集上清液,60℃減壓濃縮,然后加入乙醇至乙醇體積濃度為75%,靜置24h后離心,收集沉淀,得堿提粗糖聚合物ab4;
s5純化:利用sevag法分別將步驟s3、s4所得ab1、ab2、ab3、ab4粗糖聚合物進行除蛋白,除蛋白后粗糖聚合物用透析袋(截留分子量為1000da)進行透析、凍干,得牛膝糖聚合物(牛膝糖聚合物ab1、ab2、ab3、ab4)。
實施例2牛膝糖聚合物abp3-2
所述牛膝糖聚合物abp3-2是由實施例1所得牛膝糖聚合物ab3二次純化所得,具體方法如下:
1)離子交換柱層析:取100mg實施例1所得牛膝糖聚合物ab3,溶于5ml的去離子水中,上樣于deae-cellulose52柱,在不同鹽濃度的洗脫液條件下出現2個峰,其中峰一為水(0m的nacl)洗脫部分,峰二為0.1m的nacl洗脫部分(洗脫過程中使用苯酚-硫酸法跟蹤洗脫曲線,根據洗脫曲線分別收集糖部分),分別將所得洗脫液濃縮、冷凍干燥后,得到兩種糖聚合物(峰一糖聚合物、峰二糖聚合物);
2)分子篩凝膠層析:將上述凍干后的峰二糖聚合物樣品,用水進行溶解,離心,取上清液,上sephacryls-100hr柱,用水進行洗脫,使用苯酚-硫酸法跟蹤洗脫曲線,出現一個單一對稱峰,收集主峰,濃縮,冷凍干燥,得牛膝糖聚合物abp3-2。
實施例3牛膝糖聚合物abw1-1
所述牛膝糖聚合物abw1-1是由實施例1所得牛膝糖聚合物ab1二次純化所得,具體方法如下:
離子交換柱層析:取100mg實施例1所得牛膝糖聚合物ab1,溶于5ml的去離子水中,上樣于deae-cellulose52柱,在不同鹽濃度的洗脫液條件下出現2個峰,其中峰一為水(0m的nacl)洗脫部分,峰二為0.1m的nacl洗脫部分(洗脫過程中使用苯酚-硫酸法跟蹤洗脫曲線,根據洗脫曲線分別收集糖部分),收集峰一洗脫液,濃縮、冷凍干燥,得牛膝糖聚合物abw1-1。
實施例4牛膝糖聚合物abp1-2
所述牛膝糖聚合物abp1-2是由實施例1所得牛膝糖聚合物ab1二次純化所得,具體方法如下:
離子交換柱層析:取100mg實施例1所得牛膝糖聚合物ab1,溶于5ml的去離子水中,上樣于deae-cellulose52柱,在不同鹽濃度的洗脫液條件下出現2個峰,其中峰一為水(0m的nacl)洗脫部分,峰二為0.1m的nacl洗脫部分(洗脫過程中使用苯酚-硫酸法跟蹤洗脫曲線,根據洗脫曲線分別收集糖部分),收集峰二洗脫液,濃縮、冷凍干燥,得牛膝糖聚合物abp1-2。
實施例5牛膝糖糖聚合物abp4-3
所述牛膝糖聚合物abp4-3是由實施例1所得牛膝糖聚合物ab4二次純化所得,具體方法如下:
1)離子交換柱層析:取100mg實施例1所得牛膝糖聚合物ab4,溶于5ml的去離子水中,上樣于deae-cellulose52柱,在不同鹽濃度的洗脫液條件下出現4個峰,其中峰一為0.05m的nacl洗脫部分,峰二為0.1m的nacl洗脫部分,峰三為0.15m的nacl洗脫部分,峰四為0.2m的nacl洗脫部分(洗脫過程中使用苯酚-硫酸法跟蹤洗脫曲線,根據洗脫曲線分別收集糖部分),分別將所得洗脫液濃縮、冷凍干燥后,得到四種糖聚合物;
6)分子篩凝膠層析:將上述凍干后的峰三糖聚合物樣品,用水進行溶解,離心,取上清液,上sephacryls-100hr柱,用水進行洗脫,使用苯酚-硫酸法跟蹤洗脫曲線,出現一個單一對稱峰,收集主峰,濃縮,冷凍干燥,得牛膝糖聚合物abp4-3。
試驗例一、牛膝糖聚合物abw1-1、abp1-2、abp3-2和abp4-3的結構分析
(一)試驗材料:牛膝糖聚合物abw1-1、abp1-2、abp3-2和abp4-3。
(二)試驗方法:
1.單糖組成分析
(1)pmp柱前衍生化hplc分析
樣品處理:
精確稱取4.0mg試驗材料于具塞試管中,加入2m三氟乙酸(tfa)2.0ml,充n2封管,置于油浴鍋中在120℃油浴水解6h。冷卻至室溫,重復加甲醇旋干,去除tfa,用去離子水溶解至1ml,離心,各吸取100μl樣品溶液,加入100μl0.3m的naoh溶液,再加入100μl0.5mpmp甲醇溶液,混勻,70℃水浴鍋上反應30min,冷卻,加入105μl0.3m的hcl溶液中和,加去離子水至1ml,再加入等體積的氯仿溶液,劇烈震搖,離心,去氯仿相,如此反復2次萃取,取水相用0.45μm濾膜過濾后供hplc進樣分析。
色譜條件:
色譜柱:agilentzorbaxeclipsexdb-c18,4.6×250mm,5μm;流動相:0.1m磷酸鹽(ph=6.9)緩沖液-乙腈(v/v為83:17);檢測波長:250nm;流速:1ml/min;進樣體積:20μl。
(2)高效離子色譜分析
樣品處理:
精密稱取4.0mg試驗材料于具塞試管中加入2mtfa2ml,在120℃中恒溫油浴6h,冷卻至室溫,重復加甲醇旋干以去除tfa,用蒸餾水溶解至1ml,0.45μm濾膜過濾,供離子色譜分析。
色譜條件:
色譜柱:dionexcarbopacpa10,規格250×4mm;流動相:水-200mmnaoh溶液(v/v為92:8),流速為1.0ml/min;進樣體積為20μl,柱溫為30℃。
2.紅外光譜檢測
將干燥的試驗材料2.0mg與kbr研磨后,壓片,用perkineimerft/ir-100在4000-450cm-1范圍內進行掃描。
3.甲基化/gc-ms分析
稱取干燥的試驗材料8.0mg于反應瓶中,加入無水dmso8ml,再加入干燥的氫氧化鈉800mg,磁力攪拌10min,冰浴條件下,通入n2避光加入碘甲烷4.5ml,室溫攪拌反應8h,然后加入2ml的蒸餾水分解殘留的碘甲烷,用蒸餾水透析24h,減壓濃縮,樣品烘干后用1ml氯仿溶解。
甲基化完全后置于具塞試管中用2mol/l的tfa在120℃恒溫油浴水解6h,減壓蒸發至干,再重復多次加入甲醇旋干至ph為中性,然后用20mg的nabh4在40℃下反應30min還原水解產物。再使用100μl的冰醋酸終止反應,樣品在低壓下旋干,然后加入2ml的乙酰酐和吡啶用來乙酰化。保持95℃磁力攪拌反應2h。然后反復加甲苯3次,旋干,用1ml氯仿溶解,再用等體積的蒸餾水洗滌4次,除去水層,最后用無水硫酸鈉干燥氯仿層,再過濾除去硫酸鈉固體,減壓濃縮蒸干,供gc-ms分析。
4.核磁共振分析
取各牛膝糖聚合物樣品30mg溶于0.6mld2o中,反復多次凍干后,置于核磁管中,用500mhz核磁共振儀brukerav-500記錄其1hnmr,13cnmr,hsqc,hmbc等圖譜。
(三)試驗結果:
1.牛膝糖聚合物abp3-2結構分析
(1)單糖組成分析
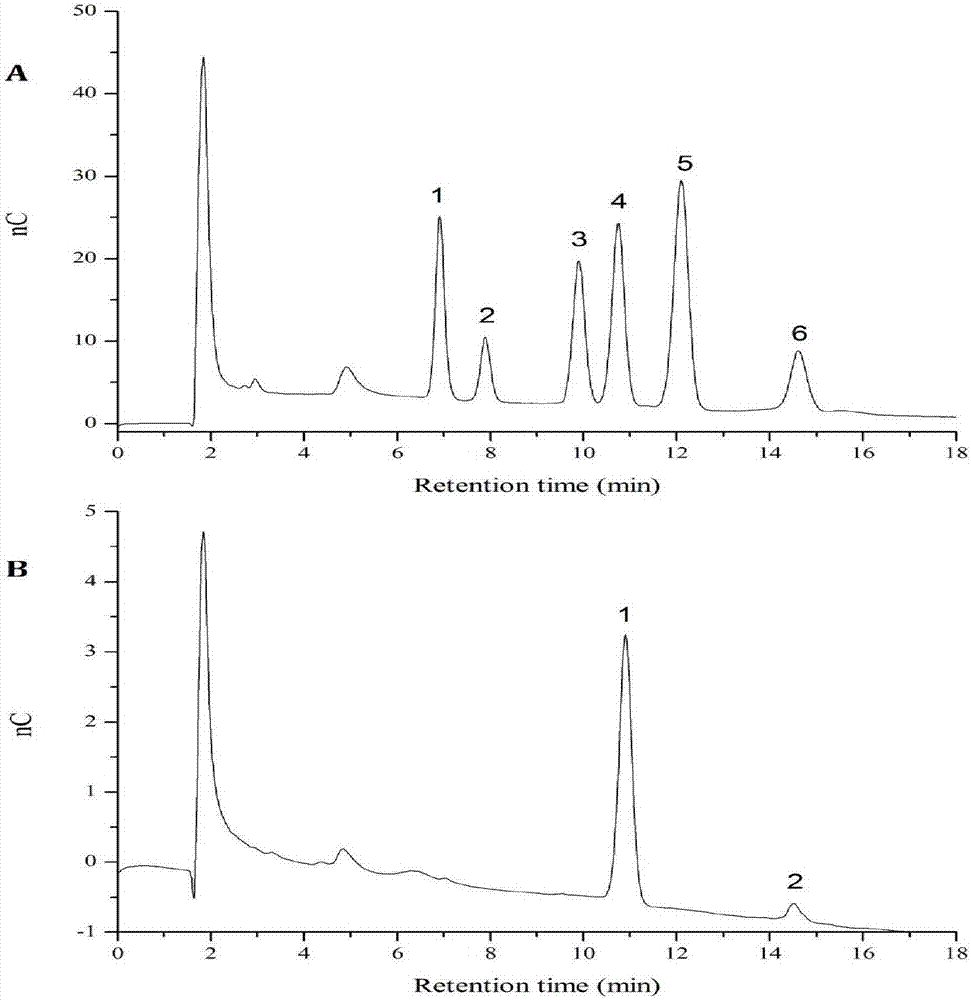
如圖1所示,由完全酸水解產物hpaec-pad圖譜可知,abp3-2是由葡萄糖和果糖組成。圖a為各單糖標準品的hpaec-pad圖譜,1:鼠李糖,2:阿拉伯糖,3:半乳糖,4:葡萄糖,5:甘露糖/木糖,6:果糖。圖b為abp3-2的hpaec-pad圖譜,1:葡萄糖,2:果糖。
(2)紅外光譜分析
如圖2所示,由abp3-2紅外光譜圖可知,abp3-2含有糖聚物的特征吸收峰:3368cm-1為o-h伸縮振動,2936cm-1為c-h伸縮振動,1642cm-1為羰基吸收峰。1740cm-1區間未見吸收峰說明abp3-2不含有糖醛酸,929cm-1和815cm-1為呋喃環的特征吸收峰;871cm-1處有吸收峰,說明abp3-2為主要由β型糖殘基組成。
(3)甲基化/gc-ms分析
abp3-2甲基化分析,經過水解,還原乙酰化后,gc-ms檢測,由gc-ms圖譜可知,abp3-2含有→1)-β-d-fruf-(2→和→2)-β-d-fruf-(6→糖殘基。
(4)核磁共振分析
本試驗通過1hnmr、13cnmr、hsqc對abp3-2的糖殘基的碳原子和氫原子的化學位移進行歸屬,然后用hmbc確認其連接順序。圖4-7為abp3-2的1hnmr、13cnmr、hsqc和hmbc圖譜。
根據圖3-6的核磁圖譜可知各個碳和氫的歸屬,如下表1所示。
表1abw3-1核磁共振分析結果
aunresolvedfromothersignals.
綜上所述:abp3-2是一種由葡萄糖和果糖組成的果聚糖,從甲基化分析說明其含有→1)-β-d-fruf-(2→和→2)-β-d-fruf-(6→糖殘基,不同糖殘基之間的連接順序由二維核磁hmbc譜圖分析得出,由以上分析得出abp3-2的結構為:
其中n為1~6或8~30。
2.牛膝糖聚合物abw1-1、abp1-2和abp4-3的結構分析
同理,分別對糖聚合物abw1-1、abp1-2、abp4-3的結構進行上述同樣的分析,并分別獲得以下信息:
(1)由圖7-12可知,牛膝糖聚合物abw1-1是由葡萄糖和果糖組成的果聚糖,主鏈由→2)-α-d-fruf-(1→組成,支鏈由→2)-β-d-fruf-(6→組成,末端糖殘基由α-d-glcp-(1→和→2)-β-d-fruf組成,abw1-1結構為:
其中x為2~30,y為2~30。
(2)由圖13-18可知,牛膝糖聚合物abp1-2是由葡萄糖和果糖組成的果聚糖,主鏈由→2)-α-d-fruf-(1→組成,支鏈由→2)-β-d-fruf-(6→組成,末端糖殘基由α-d-glcp-(1→和→2)-β-d-fruf組成,abp1-2結構為:
其中n為1~6或8~30,m為1~11或13~30。
(3)由圖19-24可知,牛膝糖聚合物abp4-3是由阿拉伯糖,半乳糖,鼠李糖和半乳糖醛酸組成的雜多糖,主鏈由→4)-α-d-galpa-(1→和→2,4)-α-d-galpa-(1→組成,支鏈包括→5)-α-l-araf-(1→、→3)-α-l-araf-(1→、→2,3,5)-α-l-araf-(1→、→6)-β-d-galp-(1→、→3,6)-β-d-galp-(1→、α-l-araf-(1→、α-l-rhap-(1→和β-d-galp-(1→,abpb-3結構為:
其中n為1~30,m為1~30,x為1~30,y為1~30。
試驗例二、牛膝糖聚合物純品的抗骨質疏松作用研究
(一)試驗材料:牛膝糖聚合物abw1-1、abp4-3。
(二)試驗對象:斑馬魚幼魚,購于上海南方模式生物科技發展有限公司,60只。
(三)試驗方法:
1.實驗設計與分組:
本發明采用斑馬魚動物模型,以糖皮質激素潑尼松龍為模型藥,建立一種快速,高效的骨質疏松模型。將受精后3天的斑馬魚胚胎暴露于25μm的潑尼松龍溶液中,28.5℃下在6孔板中培養96h,至斑馬魚頭部骨骼形成,設模型組、空白對照組、陽性對照組、給藥組。用此模型觀察牛膝糖聚合物abw1-1及abp4-3對潑尼松龍誘導的骨質疏松的防治作用。各組動物和給藥方法見表2,其中陽性藥aln為阿侖膦酸鈉,陰性對照組為0.1%dmso。造模后暴露給藥,給藥后連續培養96h。
表2實驗設計與分組
2.斑馬魚胚胎收集與培養
控制斑馬魚成魚日夜節奏,晝夜時間控制在14h:10h,收集斑馬魚受精卵,置于培養皿中,加入培養基(0.2%的速溶海鹽溶于去離子水中),置于恒溫培養箱中培養,培養溫度為28.5℃。
3.牛膝糖聚物對潑尼松龍誘導的斑馬魚骨質疏松模型的影響
將受精后3天的斑馬魚幼魚放入6孔板中,加入培養基,每孔放10條幼魚,分為陰性對照組,模型組,陽性對照組,給藥組,每組做3份。受精后第4天開始根據表2方法給藥,將6孔板密封放入28.5℃培養箱中恒溫培養96h,培養過程中斑馬魚幼魚不喂食。
4.鈣黃綠素染色
給藥結束后,將受精后7天的斑馬魚幼魚用養魚水反復沖洗三次,浸沒于0.2%的鈣黃綠素溶液中5min,然后幼魚再用養魚水反復徹底沖洗三次(5min/次),用0.016%ms-222麻醉致死,用3%的甲基纖維素固定幼魚,熒光顯微鏡下觀察。5.圖像采集以及頭部骨骼定量分析
nikonsmz1500熒光倒置顯微鏡觀察鈣黃綠素染色的斑馬魚頭骨腹面,其配套的成像軟件采集圖像,所有的圖像均采用相同的光強和曝光設置,圖像最后使用adobephotoshop7.0軟件調節處理。專業的形態分析軟件nis-elementsd3.1測定受精后7天斑馬魚幼魚頭部骨骼的相對熒光強度rfi(relativefluorescenceintensity),以反映骨密度。
(四)實驗結果
1.潑尼松龍誘導骨質疏松斑馬魚鈣黃綠素染色圖像
牛膝糖聚合物對潑尼松龍誘導的骨質疏松斑馬魚進行鈣黃綠素染色,結果見圖25、27。如圖所示,25μm潑尼松龍處理后,相對于con組,受精7天后的斑馬魚幼魚頭部骨骼的鈣黃綠素的染色面積和染色強度明顯減少,說明糖皮質激素潑尼松龍誘導斑馬魚骨質疏松模型成功。與pre組比較,牛膝糖聚合物abw1-1以及abp4-3給藥組明顯增強斑馬魚幼魚頭部骨骼的染色面積以及染色強度,說明牛膝糖聚合物abw1-1和abp4-3對潑尼松龍誘導的斑馬魚骨質疏松具有良好的治療效果。
2.潑尼松龍誘導骨質疏松斑馬魚相對熒光強度定量分析
牛膝糖聚合物對潑尼松龍誘導骨質疏松斑馬魚頭部骨骼的相對熒光強度定量分析見圖26、28。如圖所示,與con組比較,模型組斑馬魚頭部骨骼的相對熒光強度極顯著地減少,差異具有統計學意義(p<0.0001),說明潑尼松龍誘導斑馬魚骨質疏松模型成功。與pre組比較,牛膝糖聚合物abw1-1和abp4-3均可極顯著性提高斑馬魚頭部骨骼的相對熒光強度,且差異具有統計學意義(p<0.0001),說明牛膝糖聚合物abw1-1和abp4-3對潑尼松龍誘導的斑馬魚骨質疏松具有良好的治療效果。
綜上所述,本發明制備的牛膝糖聚合物純品abw1-1和abp4-3均能顯著增加糖皮質激素誘導骨質疏松斑馬魚頭部骨骼的相對熒光強度,明顯增強斑馬魚頭部骨骼鈣黃綠素染色面積以及染色強度,從而發揮其治療骨質疏松癥的作用。
- 還沒有人留言評論。精彩留言會獲得點贊!