米諾地爾分子印跡聚合物及其制備方法以及米諾地爾固相萃取柱和分離純化米諾地爾的方法與流程
本發明涉及米諾地爾檢測技術領域,是一種米諾地爾分子印跡聚合物及其制備方法以及米諾地爾固相萃取柱和使用該米諾地爾固相萃取分離純化米諾地爾的方法。
背景技術:
米諾地爾是20世紀70年代研制的一種鉀通道開放劑,主要用于治療頑固性高血壓。米諾地爾是一種二型的5a還原酶抑制劑。因為米諾地爾在高血壓的治療中發現其具有使毛發變長變粗的作用,所以目前它被開發應用作為治療男性禿發的外用藥物。到目前為止,米諾地爾引起頭發生長的機制仍然沒有完全解釋清楚,認為其中包含了多種途徑。1988年美國FDA批準米諾地爾可用于治療男性雄激素性脫發。隨著人們生活水平的提高,用藥的天然性越來越受到追捧,嚴密監測米諾地爾產品的含量對于患者用藥安全具有重要意義,而隨著科學技術的發展,米諾地爾的定量方法也由原質量標準中的分光光度法提高為高效液相色譜法,目前高效液相測定含有米諾地爾的產品中的米諾地爾需要對測定樣品進行前處理,具體為:例如:稱取0.5g(精確至0.01g)試樣于10mL干燥具塞比色管中,加入10mL質量分數為1%三氯乙酸,超聲提取10min,再渦旋振蕩提取1min,以4 000r/min離心5min,上清液待凈化,若樣品中油脂含量較高,可用質量分數為1%三氯乙酸溶液飽和的正己烷先除脂,再用SPE柱凈化,準確吸取1.0mL待凈化液,移至已活化的MCX固相萃取柱中,依次用3mL水、3mL甲醇洗滌,棄去流出液,再用10mL質量分數為5%氨化甲醇洗脫,收集洗脫液,洗脫液于50℃水浴中用氮氣吹干后,用甲醇定容至1.0mL,渦旋振蕩搖勻,過0.22μm濾膜供HPLC測定。由此可以看出,目前測定產品中的米諾地爾的方法前處理手段操作復雜,并且該前處理手段對樣品中米諾地爾的純化度不高,使得其后續采用高效液相色譜測定時,對樣品存在較大的干擾。
技術實現要素:
本發明提供了一種米諾地爾分子印跡聚合物及其制備方法,根據本發明方法得到的米諾地爾分子印跡聚合物對米諾地爾具有較高的親和性,本發明還提供了一種米諾地爾固相萃取柱以及使用該米諾地爾固相萃取柱進行萃取分離純化米諾地爾的方法,采用本發明的米諾地爾固相萃取柱進行萃取分離純化米諾地爾的方法,有效解決了目前測定市售的含有米諾地爾的生發產品中米諾地爾時前處理手段操作復雜、樣品干擾大的問題。
本發明的技術方案之一是通過以下措施來實現的:一種米諾地爾分子印跡聚合物,按下述方法得到:
第一步,首先用無機酸對球形硅膠進行回流清洗18小時至24小時得到活化后的硅膠,然后用水將活化后的硅膠洗至中性后烘干至恒重,其中,無機酸與球形硅膠的質量比為5:1至10:1;
第二步,按重量份數計將1份活化后的硅膠分散于10份至25份的有機試劑Ⅰ中得到硅膠分散溶液,接著向其中加入3-氨丙基三乙氧基硅烷對球形硅膠進行硅烷化,硅烷化的反應條件為氮氣環境下110℃至150℃反應18小時至24小時,球形硅膠與3-氨丙基三乙氧基硅烷的質量比為1:1至1:1.5,硅烷化反應完成后得到硅烷化硅膠,用有機試劑Ⅱ洗滌硅烷化硅膠除去未反應完的3-氨丙基三乙氧基硅烷,然后將硅烷化硅膠干燥至恒重;
第三步,按1g干燥后的硅烷化硅膠溶解于10ml至20ml甲醇的比例將干燥后的硅烷化硅膠溶解于甲醇中得到硅烷化硅膠甲醇溶液,按干燥后的硅烷化硅膠:4,4′-偶氮-二(4-氰基戊酸):對二甲氨基吡啶:二環己基碳二亞胺的質量比為454.5:11.36:1:8.38至454.5:12.2:1:9.8的比例向硅烷化硅膠甲醇溶液中加入4,4′-偶氮-二(4-氰基戊酸)、對二甲氨基吡啶和二環己基碳二亞胺,采用超聲混合均勻并溶解,在氮氣環境下80℃至85℃反應18小時至24小時得到表修飾硅膠混合物,將表修飾硅膠混合物進行過濾后的濾餅用有機試劑Ⅲ進行清洗除去未反應完的4,4′-偶氮-二(4-氰基戊酸)、對二甲氨基吡啶和二環己基碳二亞胺得到表面修飾硅膠,然后將表面修飾硅膠干燥至恒重;
第四步,將功能單體、交聯劑、致孔劑、引發劑、表面修飾硅膠和印跡分子米諾地爾采用超聲方式進行混合均勻并溶解得到混合溶液,其中,原料中:印跡分子米諾地爾與功能單體之間物質的量之比為0.005:1至5:1,交聯劑與功能單體物質的量之比為0.5:1至20:1,表面修飾硅膠與功能單體物質的量之比為1:1至1:1.5,致孔劑的用量為功能單體、交聯劑、表面修飾硅膠和印跡分子米諾地爾總體積的30%至75%,引發劑的用量為功能單體、交聯劑、致孔劑、表面修飾硅膠和印跡分子米諾地爾總體積的2%至4%;
第五步,將混合溶液進行聚合得到聚合物;
第六步,將聚合物先進行過濾棄去濾液得到濾餅,然后用有機試劑Ⅳ沖洗濾餅除去其中的沒有參加反應的功能單體、交聯劑和引發劑,再用有機混合溶液洗脫除去濾餅中的印跡分子米諾地爾得到無印跡分子的聚合物;
第七步,將無印跡分子的聚合物用有機試劑Ⅴ洗至中性,在真空下干燥至恒重得到米諾地爾分子印跡聚合物。
下面是對上述發明技術方案之一的進一步優化或/和改進:
上述第五步中的聚合采用熱引發聚合,熱引發聚合的溫度為60℃至90℃,聚合反應時間為6小時至48小時;或,第五步中的聚合采用光引發聚合,光引發聚合的條件為:高壓汞燈的功率為125W或150W,聚合時間為0.5小時至48小時。
上述第二步中,用有機試劑Ⅱ洗滌的具體方法為:先用苯洗滌2次至3次,再用丙酮洗滌2次至3次;或,第二步中,用有機試劑Ⅱ洗滌的具體方法為:先用甲苯洗滌2次至3次,再用乙醚洗滌2次至3次;或,第二步中,用有機試劑Ⅱ洗滌的具體方法為:先用乙酸乙酯洗滌2次至3次,再用甲醇洗滌2次至3次;或,第六步中的有機混合溶液為甲醇與乙酸按體積比9:1至4:1混合后得到的有機混合溶液;或,第六步中的有機混合溶液為乙腈與乙酸按體積比9:1至4:1混合后得到的有機混合溶液。
上述當印跡分子米諾地爾呈酸性時功能單體選用酰胺類的單體,當印跡分子米諾地爾呈堿性時功能單體選用吡啶類的單體或丙烯酸類的單體,酰胺類的單體為丙烯酰胺或二丙烯酰胺-2-甲基-1-丙磺酸,吡啶類的單體為2,6-二氨基吡啶或4-乙烯基吡啶(4-VP)或2-乙烯基吡啶,丙烯酸類單體為丙烯酸或甲基丙烯酸或三氯甲基丙烯酸或甲基丙烯酸甲酯或甲基丙烯酸羥乙基酯或甲基丙烯酸二乙胺乙基酯;或/和,交聯劑為三羥甲基丙烷三甲基丙烯酸酯或N,N-亞甲基二丙烯酰胺或乙二醇二甲基丙烯酸酯或二乙烯基苯;或/和,致孔劑為二氯甲烷、氯仿、丙酮、乙腈、甲醇、異丙醇中的一種;或/和,引發劑為有機過氧類或偶氮類化合物;或/和,第一步中,無機酸為鹽酸或硝酸,鹽酸或硝酸的體積百分數濃度為5%至10%;或/和,第二步中,有機試劑Ⅰ為苯或甲苯或乙酸乙酯;或/和,第三步中,采用有機試劑Ⅲ進行清洗時先用甲醇沖洗2次至3次,再用石油醚沖洗2次至3次;或/和,第六步中的有機試劑Ⅳ為二氯甲烷、氯仿、丙酮、乙腈、甲、異丙醇中的一種;或/和,第七步中的有機試劑Ⅴ為二氯甲烷、氯仿、丙酮、乙腈、甲醇、異丙醇中的一種。
本發明的技術方案之二是通過以下措施來實現的:一種米諾地爾分子印跡聚合物的制備方法,按下述步驟進行:第一步,首先用無機酸對球形硅膠進行回流清洗18小時至24小時得到活化后的硅膠,然后用水將活化后的硅膠洗至中性后烘干至恒重,其中,無機酸與球形硅膠的質量比為5:1至10:1,球形硅膠為300目至400目的球形硅膠;
第二步,按重量份數計將1份活化后的硅膠分散于10份至25份的有機試劑Ⅰ中得到硅膠分散溶液,接著向其中加入3-氨丙基三乙氧基硅烷對球形硅膠進行硅烷化,硅烷化的反應條件為氮氣環境下110℃至150℃反應18小時至24小時,球形硅膠與3-氨丙基三乙氧基硅烷的質量比為1:1至1:1.5,硅烷化反應完成后得到硅烷化硅膠,用有機試劑Ⅱ洗滌硅烷化硅膠除去未反應完的3-氨丙基三乙氧基硅烷,然后將硅烷化硅膠干燥至恒重;
第三步,按1g干燥后的硅烷化硅膠溶解于10ml至20ml甲醇的比例將干燥后的硅烷化硅膠溶解于甲醇中得到硅烷化硅膠甲醇溶液,按干燥后的硅烷化硅膠:4,4′-偶氮-二(4-氰基戊酸):對二甲氨基吡啶:二環己基碳二亞胺的質量比為454.5:11.36:1:8.38至454.5:12.2:1:9.8的比例向硅烷化硅膠甲醇溶液中加入4,4′-偶氮-二(4-氰基戊酸)、對二甲氨基吡啶和二環己基碳二亞胺,采用超聲混合均勻并溶解,在氮氣環境下80℃至85℃反應18小時至24小時得到表修飾硅膠混合物,將表修飾硅膠混合物進行過濾后的濾餅用有機試劑Ⅲ進行清洗除去未反應完的4,4′-偶氮-二(4-氰基戊酸)、對二甲氨基吡啶和二環己基碳二亞胺得到表面修飾硅膠,然后將表面修飾硅膠干燥至恒重;
第四步,將功能單體、交聯劑、致孔劑、引發劑、表面修飾硅膠和印跡分子米諾地爾采用超聲方式進行混合均勻并溶解得到混合溶液,其中,原料中:印跡分子米諾地爾與功能單體之間物質的量之比為0.005:1至5:1,交聯劑與功能單體物質的量之比為0.5:1至20:1,表面修飾硅膠與功能單體物質的量之比為1:1至1:1.5,致孔劑的用量為功能單體、交聯劑、表面修飾硅膠和印跡分子米諾地爾總體積的30%至75%,引發劑的用量為功能單體、交聯劑、致孔劑、表面修飾硅膠和印跡分子米諾地爾總體積的2%至4%;
第五步,將混合溶液進行聚合得到聚合物;
第六步,將聚合物先進行過濾棄去濾液得到濾餅,然后用有機試劑Ⅳ沖洗濾餅除去其中的沒有參加反應的功能單體、致孔劑、交聯劑和引發劑,再用有機混合溶液洗脫除去濾餅中的印跡分子米諾地爾得到無印跡分子的聚合物;
第七步,將無印跡分子的聚合物用有機試劑Ⅴ洗至中性,在真空下干燥至恒重得到米諾地爾分子印跡聚合物。
下面是對上述發明技術方案之二的進一步優化或/和改進:
上述第五步中的聚合采用熱引發聚合,熱引發聚合的溫度為60℃至90℃,聚合反應時間為6小時至48小時;或,第五步中的聚合采用光引發聚合,光引發聚合的條件為:高壓汞燈的功率為125W或150W,聚合時間為0.5小時至48小時。
上述第二步中,用有機試劑Ⅱ洗滌的具體方法為:先用苯洗滌2次至3次,再用丙酮洗滌2次至3次;或,第二步中,用有機試劑Ⅱ洗滌的具體方法為:先用甲苯洗滌2次至3次,再用乙醚洗滌2次至3次;或,第二步中,用有機試劑Ⅱ洗滌的具體方法為:先用乙酸乙酯洗滌2次至3次,再用甲醇洗滌2次至3次;或,第六步中的有機混合溶液為甲醇與乙酸按體積比9:1至4:1混合后得到的有機混合溶液;或,第六步中的有機混合溶液為乙腈與乙酸按體積比9:1至4:1混合后得到的有機混合溶液。
上述當印跡分子米諾地爾呈酸性時功能單體選用酰胺類的單體,當印跡分子米諾地爾呈堿性時功能單體選用吡啶類的單體或丙烯酸類的單體,酰胺類的單體為丙烯酰胺或二丙烯酰胺-2-甲基-1-丙磺酸,吡啶類的單體為2,6-二氨基吡啶或4-乙烯基吡啶(4-VP)或2-乙烯基吡啶,丙烯酸類單體為丙烯酸或甲基丙烯酸或三氯甲基丙烯酸或甲基丙烯酸甲酯或甲基丙烯酸羥乙基酯或甲基丙烯酸二乙胺乙基酯;或/和,交聯劑為三羥甲基丙烷三甲基丙烯酸酯或N,N-亞甲基二丙烯酰胺或乙二醇二甲基丙烯酸酯或二乙烯基苯;或/和,致孔劑為二氯甲烷、氯仿、丙酮、乙腈、甲醇、異丙醇中的一種;或/和,引發劑為有機過氧類或偶氮類化合物;或/和,第一步中,無機酸為鹽酸或硝酸,鹽酸或硝酸的體積百分數濃度為5%至10%;或/和,第二步中,有機試劑Ⅰ為苯或甲苯或乙酸乙酯;或/和,第三步中,采用有機試劑Ⅲ進行清洗時先用甲醇沖洗2次至3次,再用石油醚沖洗2次至3次;或/和,第六步中的有機試劑Ⅳ為二氯甲烷、氯仿、丙酮、乙腈、甲、異丙醇中的一種;或/和,第七步中的有機試劑Ⅴ為二氯甲烷、氯仿、丙酮、乙腈、甲醇、異丙醇中的一種。
本發明的技術方案之三是通過以下措施來實現的:一種使用米諾地爾分子印跡聚合物得到的米諾地爾固相萃取柱,按下述方法得到:將米諾地爾分子印跡聚合物填充到注射器中制得米諾地爾固相萃取柱。
本發明的技術方案之四是通過以下措施來實現的:一種使用米諾地爾固相萃取柱進行分離純化米諾地爾的方法,按下述方法進行:第一步,按體積份數計,分別取1份至2份市售含有米諾地爾的生發產品、3份至5份的水、3份至5份的甲醇、3份至5份的甲醇和乙酸按體積比9:1混合的混合洗脫液;第二步,按1.5g至2g米諾地爾分子印跡聚合物處理1mL至2mL市售含有米諾地爾的生發產品計,選擇合適規模的米諾地爾固相萃取柱;第三步,將市售含有米諾地爾的生發產品加入到米諾地爾固相萃取柱中,先依次用水和甲醇進行沖洗,再用混合洗脫液進行洗脫,收集洗脫成分。
本發明方法得到的米諾地爾分子印跡聚合物對米諾地爾具有較高的親和性,采用本發明的米諾地爾分子印跡聚合物制備得到的米諾地爾固相萃取柱進行萃取分離純化米諾地爾的方法,使得測定市售的含有米諾地爾的生發產品中米諾地爾時前處理手段操作簡單,便于實施,并且,采用對米諾地爾具有較高親和性的米諾地爾分子印跡聚合物分離純化待測樣品,有效提高待測樣品的純度,有效減少高效液相色譜測定待測樣品時對樣品的干擾。
附圖說明
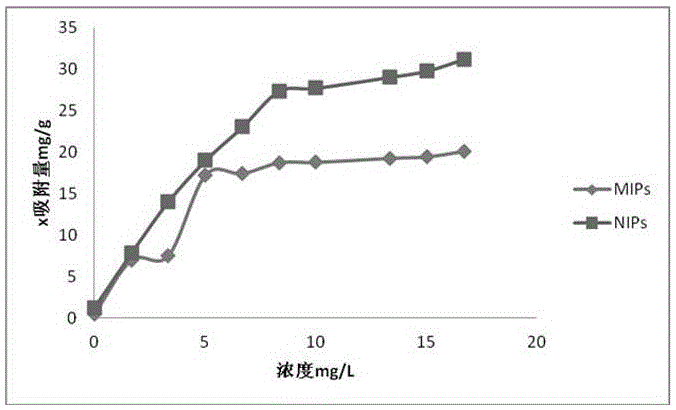
附圖1為本發明溶液中米諾地爾的濃度變化與單位質量聚合物微球對底物的吸附量的關系曲線圖。
具體實施方式
本發明不受下述實施例的限制,可根據本發明的技術方案與實際情況來確定具體的實施方式。本發明中所提到各種化學試劑和化學用品如無特殊說明,均為現技術中公知公用的化學試劑和化學用品;本發明中的百分數如沒有特殊說明,均為質量百分數;本發明中的水如沒有特殊說明,為自來水或純凈水;本發明中的溶液若沒有特殊說明,均為溶劑為水的水溶液,例如,鹽酸溶液即為鹽酸水溶液;本發明中的常溫一般指15℃到25℃的溫度,一般定義為25℃。
下面結合實施例及附圖對本發明作進一步描述:
實施例1,該米諾地爾分子印跡聚合物,按下述制備方法得到:
第一步,首先用無機酸對球形硅膠進行回流清洗18小時至24小時得到活化后的硅膠,然后用水將活化后的硅膠洗至中性后烘干至恒重,其中,無機酸與球形硅膠的質量比為5:1至10:1,球形硅膠為300目至400目的球形硅膠;
第二步,按重量份數計將1份活化后的硅膠分散于10份至25份的有機試劑Ⅰ中得到硅膠分散溶液,接著向其中加入3-氨丙基三乙氧基硅烷對球形硅膠進行硅烷化,硅烷化的反應條件為氮氣環境下110℃至150℃反應18小時至24小時,球形硅膠與3-氨丙基三乙氧基硅烷的質量比為1:1至1:1.5,硅烷化反應完成后得到硅烷化硅膠,用有機試劑Ⅱ洗滌硅烷化硅膠除去未反應完的3-氨丙基三乙氧基硅烷,然后將硅烷化硅膠干燥至恒重;
第三步,按1g干燥后的硅烷化硅膠溶解于10ml至20ml甲醇的比例將干燥后的硅烷化硅膠溶解于甲醇中得到硅烷化硅膠甲醇溶液,按干燥后的硅烷化硅膠:4,4′-偶氮-二(4-氰基戊酸):對二甲氨基吡啶:二環己基碳二亞胺的質量比為454.5:11.36:1:8.38至454.5:12.2:1:9.8的比例向硅烷化硅膠甲醇溶液中加入4,4′-偶氮-二(4-氰基戊酸)、對二甲氨基吡啶和二環己基碳二亞胺,采用超聲混合均勻并溶解,在氮氣環境下80℃至85℃反應18小時至24小時得到表修飾硅膠混合物,將表修飾硅膠混合物進行過濾后的濾餅用有機試劑Ⅲ進行清洗除去未反應完的4,4′-偶氮-二(4-氰基戊酸)、對二甲氨基吡啶和二環己基碳二亞胺得到表面修飾硅膠,然后將表面修飾硅膠干燥至恒重;
第四步,將功能單體、交聯劑、致孔劑、引發劑、表面修飾硅膠和印跡分子米諾地爾采用超聲方式進行混合均勻并溶解得到混合溶液,其中,原料中:印跡分子米諾地爾與功能單體之間物質的量之比為0.005:1至5:1,交聯劑與功能單體物質的量之比為0.5:1至20:1,表面修飾硅膠與功能單體物質的量之比為1:1至1:1.5,致孔劑的用量為功能單體、交聯劑、表面修飾硅膠和印跡分子米諾地爾總體積的30%至75%,引發劑的用量為功能單體、交聯劑、致孔劑、表面修飾硅膠和印跡分子米諾地爾總體積的2%至4%;
第五步,將混合溶液進行聚合得到聚合物;
第六步,將聚合物先進行過濾棄去濾液得到濾餅,然后用有機試劑Ⅳ沖洗濾餅除去其中的沒有參加反應的功能單體、致孔劑、交聯劑和引發劑,再用有機混合溶液洗脫除去濾餅中的印跡分子米諾地爾得到無印跡分子的聚合物;
第七步,將無印跡分子的聚合物用有機試劑Ⅴ洗至中性,在真空下干燥至恒重得到米諾地爾分子印跡聚合物。根據本發明方法得到的米諾地爾分子印跡聚合物對米諾地爾具有較高的親和性。
實施例2,該米諾地爾分子印跡聚合物按下述制備方法得到:第一步,首先用無機酸對球形硅膠進行回流清洗18小時或24小時得到活化后的硅膠,然后用水將活化后的硅膠洗至中性后烘干至恒重,其中,無機酸與球形硅膠的質量比為5:1或10:1,球形硅膠為300目或400目的球形硅膠;
第二步,按重量份數計將1份活化后的硅膠分散于10份或25份的有機試劑Ⅰ中得到硅膠分散溶液,接著向其中加入3-氨丙基三乙氧基硅烷對球形硅膠進行硅烷化,硅烷化的反應條件為氮氣環境下110℃或150℃反應18小時或24小時,球形硅膠與3-氨丙基三乙氧基硅烷的質量比為1:1或1:1.5,硅烷化反應完成后得到硅烷化硅膠,用有機試劑Ⅱ洗滌硅烷化硅膠除去未反應完的3-氨丙基三乙氧基硅烷,然后將硅烷化硅膠干燥至恒重;
第三步,按1g干燥后的硅烷化硅膠溶解于10ml或20ml甲醇的比例將干燥后的硅烷化硅膠溶解于甲醇中得到硅烷化硅膠甲醇溶液,按干燥后的硅烷化硅膠:4,4′-偶氮-二(4-氰基戊酸):對二甲氨基吡啶:二環己基碳二亞胺的質量比為454.5:11.36:1:8.38或454.5:12.2:1:9.8的比例向硅烷化硅膠甲醇溶液中加入4,4′-偶氮-二(4-氰基戊酸)、對二甲氨基吡啶和二環己基碳二亞胺,采用超聲混合均勻并溶解,在氮氣環境下80℃-85℃反應18小時或24小時得到表修飾硅膠混合物,將表修飾硅膠混合物進行過濾后的濾餅用有機試劑Ⅲ進行清洗除去未反應完的4,4′-偶氮-二(4-氰基戊酸)、對二甲氨基吡啶和二環己基碳二亞胺得到表面修飾硅膠,然后將表面修飾硅膠干燥至恒重;
第四步,將功能單體、交聯劑、致孔劑、引發劑、表面修飾硅膠和印跡分子米諾地爾采用超聲方式進行混合均勻并溶解得到混合溶液,其中,原料中:印跡分子米諾地爾與功能單體之間物質的量之比為0.005:1或5:1,交聯劑與功能單體物質的量之比為0.5:1或20:1,表面修飾硅膠與功能單體物質的量之比為1:1或1:1.5,致孔劑的用量為功能單體、交聯劑、表面修飾硅膠和印跡分子米諾地爾總體積的30%或75%,引發劑的用量為功能單體、交聯劑、致孔劑、表面修飾硅膠和印跡分子米諾地爾總體積的2%或4%;
第五步,將混合溶液進行聚合得到聚合物;
第六步,將聚合物先進行過濾棄去濾液得到濾餅,然后用有機試劑Ⅳ沖洗濾餅除去其中的沒有參加反應的功能單體、致孔劑、交聯劑和引發劑,再用有機混合溶液洗脫除去濾餅中的印跡分子米諾地爾得到無印跡分子的聚合物;
第七步,將無印跡分子的聚合物用有機試劑Ⅴ洗至中性,在真空下干燥至恒重得到米諾地爾分子印跡聚合物。
實施例3,作為上述實施例的優化,第五步中的聚合采用熱引發聚合,熱引發聚合的溫度為60℃至90℃,聚合反應時間為6小時至48小時;或,第五步中的聚合采用光引發聚合,光引發聚合的條件為:高壓汞燈的功率為125W或150W,聚合時間為0.5小時至48小時。
實施例4,作為上述實施例的優化,用有機試劑Ⅱ洗滌的具體方法為:先用苯洗滌2次至3次,再用丙酮洗滌2次至3次;或,第二步中,用有機試劑Ⅱ洗滌的具體方法為:先用甲苯洗滌2次至3次,再用乙醚洗滌2次至3次;或,第二步中,用有機試劑Ⅱ洗滌的具體方法為:先用乙酸乙酯洗滌2次至3次,再用甲醇洗滌2次至3次;或,第六步中的有機混合溶液為甲醇與乙酸按體積比9:1至4:1混合后得到的有機混合溶液;或,第六步中的有機混合溶液為乙腈與乙酸按體積比9:1至4:1混合后得到的有機混合溶液。
實施例5,作為上述實施例的優化,當印跡分子呈酸性時功能單體選用酰胺類的單體,當印跡分子呈堿性時選用吡啶類的單體或丙烯酸類的單體,酰胺類的單體為丙烯酰胺或二丙烯酰胺-2-甲基-1-丙磺酸,吡啶類的單體為2,6-二氨基吡啶或4-乙烯基吡啶(4-VP)或2-乙烯基吡啶,丙烯酸類為丙烯酸或甲基丙烯酸或三氯甲基丙烯酸或甲基丙烯酸甲酯或甲基丙烯酸羥乙基酯或甲基丙烯酸二乙胺乙基酯;或/和,交聯劑為三羥甲基丙烷三甲基丙烯酸酯或N,N-亞甲基二丙烯酰胺或乙二醇二甲基丙烯酸酯或二乙烯基苯;或/和,致孔劑為二氯甲烷、氯仿、丙酮、乙腈、甲醇、異丙醇中的一種;或/和,引發劑為有機過氧類或偶氮類化合物;或/和,第一步中,無機酸為鹽酸或硝酸,鹽酸或硝酸的體積百分數濃度為5%至10%;或/和,第二步中,有機試劑Ⅰ為苯或甲苯或乙酸乙酯;或/和,第三步中,采用有機試劑Ⅲ進行清洗先用甲醇沖洗2次至3次,再用石油醚沖洗2次至3次;或/和,第六步中的有機試劑Ⅳ為二氯甲烷、氯仿、丙酮、乙腈、甲醇、異丙醇中的一種;或/和,第七步中的有機試劑Ⅴ為二氯甲烷、氯仿、丙酮、乙腈、甲醇、異丙醇中的一種。
實施例6,該使用米諾地爾分子印跡聚合物得到的米諾地爾固相萃取柱,按下述方法得到:將米諾地爾分子印跡聚合物填充到注射器中制得米諾地爾固相萃取柱。
實施例7,該使用米諾地爾固相萃取柱進行分離純化米諾地爾的方法,按下述方法進行:第一步,按體積份數計,分別取1份至2份市售含有米諾地爾的生發產品、3份至5份的水、3份至5份的甲醇、3份至5份的甲醇和乙酸按體積比9:1混合的混合洗脫液;第二步,按1.5g至2g米諾地爾分子印跡聚合物處理1mL至2mL市售含有米諾地爾的生發產品計,選擇合適規模的米諾地爾固相萃取柱;第三步,將市售含有米諾地爾的生發產品加入到米諾地爾固相萃取柱中,先依次用水和甲醇進行沖洗,再用混合洗脫液進行洗脫,收集洗脫成分。采用本發明的米諾地爾固相萃取柱進行萃取分離純化米諾地爾的方法,使得測定市售的含有米諾地爾的生發產品中米諾地爾是前處理手段操作簡單,便于實施,并且,采用對米諾地爾具有較高親和性的米諾地爾分子印跡聚合物分離純化待測樣品,有效提高待測樣品的純度,有效減少高效液相色譜測定待測樣品時對樣品的干擾。
將采用本發明上述實施例得到的米諾地爾分子印跡聚合物作為MIPs(P1);采用本發明上述實施例的方法,但不添加印跡分子米諾地爾,制備空白印跡分子聚合物NIPs(P2)。
稱取一組等量的MIPs(P1)和NIPs(P2)各30mg,分別置于離心管中,加入0mmol/L至2mmol/L米諾地爾乙腈溶液5mL,于恒溫震蕩器上20℃180rpm振蕩24h,然后9000r/min離心10min,取吸附液稀釋至一定體積,用紫外分光光度計測定平衡吸附液中米諾地爾的濃度,根據結合前后溶液中米諾地爾的濃度變化計算單位質量聚合物微球對底物的吸附量,見圖1。由圖1可以看出,MIPs(P1)的吸附量明顯高于NIPs(P2)對米諾地爾的吸附性能,說明MIPs(P1)具有較高的親和性。
以上技術特征構成了本發明的實施例,其具有較強的適應性和實施效果,可根據實際需要增減非必要的技術特征,來滿足不同情況的需求。
- 還沒有人留言評論。精彩留言會獲得點贊!