一種使用相轉移催化劑合成含有哌啶基團的硅烷的方法與流程
本發明涉及一種使用相轉移催化劑合成含有哌啶基團的硅烷的方法。
技術背景
常規的織物整理劑n-(β-氨乙基)-γ-氨丙基甲基二甲氧基硅烷在處理織物時白度值較低且后期極易黃變。
技術實現要素:
本發明其目的就在于提供了一種使用相轉移催化劑合成含有哌啶基團的硅烷的方法,解決了常規的織物整理劑在處理織物時存在白度值較低且后期極易黃變的問題。
一種使用相轉移催化劑合成含哌啶基團的硅烷的方法,其特征在于,該方法包括以下步驟:首先在氮氣保護裝置中,將1,2,2,6,6-五甲基哌啶醇溶于溶劑中,加入相轉移催化劑和縛酸劑,攪拌下加熱至80~100℃,再向其中滴加氯丙基甲基二烷氧基硅烷,保溫反應3~5h,冷卻降溫,所得反應液除去生成的鹽,回收溶劑,減壓蒸餾提純含哌啶基團的硅烷。
所述溶劑的用量為1,2,2,6,6-五甲基哌啶醇質量的20-60%;
所述相轉移催化劑為烷基季銨鹽或季磷鹽如四乙基氯化銨、四丙基氯化銨、四丁基溴化銨、四丁基碘化銨、芐基三乙基氯化銨等中的一種或幾種的混合物;用量為1,2,2,6,6-五甲基哌啶醇摩爾用量的2~3%。
所述縛酸劑為常規胺類物質如乙二胺、三乙胺、三丁胺等中的一種或幾種的混合物;用量為氯丙基甲基二烷氧基硅烷摩爾用量的1.5~2.5倍。
所述氯丙基甲基二烷氧基硅烷為氯丙基甲基二甲氧基硅烷或氯丙基甲基二乙氧基硅氧烷;用量為1,2,2,6,6-五甲基哌啶醇摩爾用量的0.9~1倍。
有益效果
與現有技術相比本發明具有以下優點。
1.合成方法簡單,易于操作,反應所用氯化氫吸收劑和催化劑可回收或反復利用;
2.合成方法綠色環保,能耗低,符合綠色化學的要求,反應轉化率高且對空氣不敏感,適用于大規模生產;
3.合成的1,2,2,6,6-五甲基哌啶-4-氧基硅烷化合物可作為制備織物整理用氨基硅油的原料,在硅油中引入1,2,2,6,6-五甲基哌啶-4-氧基團能克服當前織物整理用氨基硅油普遍存在的不耐黃變的弊端。
附圖說明
以下結合附圖對本發明作進一步詳述。
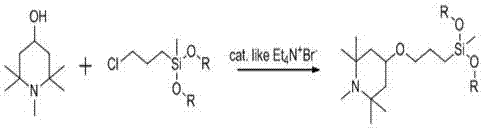
圖1為本發明中硅烷化合物的合成路徑圖。
具體實施方案
一種使用相轉移催化劑合成含哌啶基團的硅烷的方法,如圖1所示,該方法包括以下步驟:首先在氮氣保護裝置中,將1,2,2,6,6-五甲基哌啶醇溶于溶劑中,加入相轉移催化劑和縛酸劑,攪拌下加熱至80~100℃,再向其中滴加氯丙基甲基二烷氧基硅烷,保溫反應3~5h,冷卻降溫,所得反應液除去生成的鹽,回收溶劑,減壓蒸餾提純含哌啶基團的硅烷。
所述縛酸劑于1,2,2,6,6-五甲基哌啶醇與氯丙基甲基二烷氧基硅烷反應前加入或者在1,2,2,6,6-五甲基哌啶醇與氯丙基甲基二烷氧基硅烷反應降溫后加入,加入的縛酸劑不能與原料氯丙基甲基二烷氧基硅烷反應,因此不能在反應前加入乙二胺伯胺或仲胺類物質但可加入三乙胺叔胺類物質。
所述反應溶劑為與氯丙基甲基二烷氧基硅烷中烷氧基對應的低級醇或非極性溶劑dmf。
所述催化劑為烷基季銨鹽或季磷鹽,如四乙基氯化銨、四丙基氯化銨、四丁基溴化銨、四丁基碘化銨、芐基三乙基氯化銨中的一種或幾種的混合物,但不局限于此。
所述縛酸劑為無水堿性物質多常規胺類物質如乙二胺、三乙胺、三丁胺中的一種或幾種的混合物,需要注意的是si-or結構對水比較敏感,因此需要注意反應過程中不接觸水,因此不適宜用氫氧化鈉在反應過程中會產生水的物質。
所述氯丙基甲基二烷氧基硅烷中的烷氧基為甲基、乙基、異丙氧基、丁基。
所述溶劑的用量為1,2,2,6,6-五甲基哌啶醇質量的20-60%;
所述相轉移催化劑為烷基季銨鹽或季磷鹽如四乙基氯化銨、四丙基氯化銨、四丁基溴化銨、四丁基碘化銨、芐基三乙基氯化銨等中的一種或幾種的混合物;用量為1,2,2,6,6-五甲基哌啶醇摩爾用量的2~3%。
所述縛酸劑為常規胺類物質如乙二胺、三乙胺、三丁胺等中的一種或幾種的混合物;用量為氯丙基甲基二烷氧基硅烷摩爾用量的1.5~2.5倍。
所述氯丙基甲基二烷氧基硅烷為氯丙基甲基二甲氧基硅烷或氯丙基甲基二乙氧基硅氧烷;用量為1,2,2,6,6-五甲基哌啶醇摩爾用量的0.9~1倍。
實施例1:
在裝有氮氣保護裝置的三口燒瓶中加入342.6g1,2,2,6,6-五甲基哌啶醇(2mol)、150g的dmf及12g四丁基溴化銨,攪拌升溫至80℃,于恒壓滴液漏斗中滴加365.44g(2mol)氯丙基甲基二甲氧基硅烷,氣相色譜監測反應進度,至5小時后,未反應的氯丙基甲基二甲氧基硅烷小于1%,停止加熱,稍冷卻后加入270g乙二胺置換吸收氯化氫,減壓除去dmf和過量的乙二胺,冷卻分出下層粘稠的乙二胺鹽酸鹽,將上層減壓精餾,收集2mmhg壓力,140±2℃的餾分,得淺黃色液體為產品,gc檢測含量97.7%,收率93%。
實施例2:
將179.86g(1.05mol)1,2,2,6,6-五甲基哌啶醇、110g的dmf、200g三乙胺、3.3g四乙基氯化銨混合,氮氣保護下升溫至90℃,于恒壓滴液漏斗中滴加182.72g(1mol)氯丙基甲基二甲氧基硅烷,加完保溫至4h后,剩余未反應的氯丙基甲基二甲氧基硅烷小于1%,停止加熱,冷卻至室溫,過濾,用dmf清洗三乙胺鹽酸鹽,再次過濾,將所得濾液與洗液合并加熱蒸餾回收dmf和過量的三乙胺,加入活性炭70-80℃在氮氣氣氛下脫色1h,過濾,得淺黃色液體,gc檢測含量為89.8%,收率94.2%。
實施例3:
將188.43g(1.1mol)1,2,2,6,6-五甲基哌啶醇、110g的dmf、180g三乙胺、4.5g四丙基氯化銨混合,氮氣保護下升溫至90℃,于恒壓滴液漏斗中滴加210.77g(1mol)氯丙基甲基二乙氧基硅烷,加完保溫至4h后,剩余未反應的氯丙基甲基二乙氧基硅烷小于0.5%,停止加熱,冷卻至室溫,過濾,用dmf清洗三乙胺鹽酸鹽,再次過濾,將所得濾液與洗液合并加熱蒸餾回收dmf和過量的三乙胺,收集2mmhg壓力,140±2℃餾分,得淺黃色液體,gc檢測含量為97.5%,收率95.5%。
實施例4:
將188.43g(1.1mol)1,2,2,6,6-五甲基哌啶醇、110g的dmf、220g三乙胺、4.5g芐基三乙基氯化銨混合,氮氣保護下升溫至100℃,于恒壓滴液漏斗中滴加210.77g(1mol)氯丙基甲基二乙氧基硅烷,至3小時后,剩余未反應的氯丙基甲基二乙氧基硅烷小于1%,停止加熱,冷卻至室溫,過濾,用dmf清洗三乙胺鹽酸鹽,再次過濾,將所得濾液與洗液合并加熱蒸餾回收dmf和過量的三乙胺,降溫冷卻,加活性炭與硅藻土過濾,得淺黃色液體,gc檢測含量為88.7%,收率91.4%。
實施例5:
將188.43g(1.1mol)1,2,2,6,6-五甲基哌啶醇、150g乙醇、6g四丁基氯化銨及375g三丁胺混合,氮氣保護下升溫至100℃,于恒壓滴液漏斗中滴入210.77g(1mol)氯丙基甲基二乙氧基硅烷,至5小時后,剩余未反應的氯丙基甲基二乙氧基硅烷小于1%,停止加熱,冷卻至室溫,過濾,用乙醇清洗三丁胺鹽酸鹽,再次過濾,將所得濾液與洗液合并加熱蒸餾回收乙醇和過量的三丁胺,降溫冷卻,加入活性炭于80℃-90℃在氮氣氣氛下脫色1小時,加硅藻土助濾得到近無色透明的液體。gc檢測含量86.5%,收率89%。
所述提純含哌啶基團的硅烷在部分實施例中不進行減壓精餾提純,所得硅烷化合物含哌啶基團即使反應過程中通氮氣保護,仍存在黃變的情況,可在部分實施例中對蒸餾前的粗品或蒸餾后產品加硅藻土、活性炭等脫色處理。
- 還沒有人留言評論。精彩留言會獲得點贊!
- 全細胞催化合成L-2-哌啶甲酸的方法與流程
- 抗血小板聚集藥物哌啶并吡啶并4,5?二氫?1,2,3?三氮唑?鈣配合物的制備方法與流程
- 一種抗肝炎藥物的哌啶并吡啶并吡唑?鋅配合物的制備方法與流程
- 具有藥物活性的哌啶并氯代吡啶?鈣配合物的制備方法與流程
- 具有PVC穩定劑作用的哌啶并吡啶酮?錫配合物的制備方法與流程
- 具有PVC穩定劑作用的哌啶并吡啶并1,2,3?三氮唑?錫配合物的制備方法與流程
- 具有藥物活性的哌啶并吡啶?鈣配合物的制備方法與流程
- 具有PVC穩定劑功能的哌啶并吡啶酮?錫配合物的制備方法與流程
- 具有抗腫瘤活性的哌啶并吡啶類化合物的制備方法與流程
- 一種抗肝炎藥物哌啶并吡啶并1,2?二氮雜環丁烷?鋅配合物的制備方法與流程