一種錸聚合離子液體催化烯烴環氧化的方法與流程
本發明屬于化工催化領域,尤其涉及一種以錸聚合離子液體催化烯烴環氧化的方法。
背景技術:
離子液體具有很寬的溫度范圍,常溫下大多是以液態狀態存在,熱穩定性高,化學性質穩定并且有比較寬的電化學穩定電位窗口;蒸汽壓很低,不易揮發,不易燃燒,在反應中不易浪費以及造成環境污染,并有“綠色溶劑”的美譽。稀散元素化學研究所房大維等合成了陽離子為咪唑基的錸功能離子液體,這種離子液體既可以做溶劑也可用作催化劑,雙氧水為氧化劑,在烯烴環氧化反應中催化效果很好,可以循環使用催化體系,八次循環反應后的產率依舊可以達到98-99%,表明氧化條件下的錸離子液體有很好的穩定性,并且對鏈狀烯烴環氧化反應也有著高效催化活性,聚合離子液體具有著離子液體特征功能基團,當然也就有著相應特征功能。這種功能型離子液體除了可以充當一些反應媒介外,還是一種新型優良的催化劑。并且相比于負載型催化劑和凝膠型負載催化劑,這種功能型聚合離子液體催化劑既提高了動力學活性,還增大了催化比表面積。許多化學反應里,催化劑在參與催化化學反應的同時自身其實也發生了結構變化,也就是活性狀態,這種活性狀態結構是不穩定的甚至與配體分離,形成一個新的活性物種而且這類新的活性物種往往是可溶性的,不可能采用簡單的方法將產物與催化劑分離。聚合離子液體作為一種新型的催化劑,對反應增強了穩定性和改善了加工優勢,功能型錸聚合離子液體作催化劑,進行非均相催化烯烴環氧化反應,反應條件溫和,催化產率較高,選擇性,多相體系有利于催化劑的回收利用,也有利于產品的分離純化。催化劑重復反應多次后,依然能夠保持較高活性。目前,一些低級的烯烴環氧化反應(如環氧乙烷、環氧丙烷等)國內市場已經能夠大規模生產。而高級烯烴的環氧化物,還是得依賴進口,價格昂貴。無論是脂肪族環氧化物還是芳香族環氧化物,國內外的需求量都很大。所以研究聚合離子液體催化烯烴環氧化方法,有著實用價值,而且也具有廣闊的應用前景。
技術實現要素:
為了解決以上問題,本發明的目的在于提供一種錸聚合離子液體催化烯烴環氧化的方法,成本低,產率高,不污染環境,即可進行非均相催化高級烯烴環氧化反應,克服了催化劑在參與催化反應的同時自身結構變化的難題,與此同時,高級烯烴的環氧化物催化在國內就可進行,無需依賴國外進口,催化劑可回收和循環利用,有利于產品的分離純化的一種功能型錸聚合離子液體。
本發明采用的技術方案為:
一種錸聚合離子液體催化烯烴環氧化的方法,將反應物:催化劑錸聚合離子液體、溶劑、氧化劑以及底物烯烴按照一定的先后順序加入到反應釜中;于40~80℃下攪拌反應2小時后,回流冷卻反應3~10h,即完成烯烴的環氧化反應。
所述的方法,錸聚合離子液體的用量是加入反應釜中總反應物摩爾數的20%~70%。
所述的方法,所述反應物中,氧化劑的摩爾數為烯烴摩爾數的1.0~3.0倍;所述氧化劑為過氧化脲。
所述的方法,所述反應物中,所述溶劑與催化劑質量比為1:10~30;所述溶劑為1,4-二氧六環。
所述的方法,所述攪拌是采用磁力攪拌。
所述的方法,所述的反應釜為玻璃催化反應裝置。
所述的方法,所述催化劑錸聚合離子液體為1-乙烯基-3-烷基咪唑錸聚合離子液體,具體為1-乙烯基-3-丁基咪唑錸聚合離子液體P[BVIm][ReO4],1-乙烯基-3-戊基咪唑錸聚合離子液體P[PVIm][ReO4],1-乙烯基-3-己基咪唑錸聚合離子液體P[HVIm][ReO4]中一種或兩種以上混合。
所述的方法,所述1-乙烯基-3-烷基咪唑錸聚合離子液體的制備為:首先向N-乙烯基咪唑中,加入溴代烷烴,升溫至70~85℃反應5~15h后;冷卻至室溫,將產物放置于制冷機冷凍5~20h后取出,于85℃蒸發后,加入乙酸乙酯和乙腈的混合溶劑將結晶物溶解,再于85℃下蒸發重結晶,反復操作2-3次,再在70℃下真空干燥處理12~36h,得到紅棕色粘稠液體產品備用;準備一條陰離子交換樹脂柱,對陰離子交換樹脂除雜,把處理過的陰離子交換樹脂裝柱,裝入長100cm,內徑6cm離子交換柱中。然后用堿性溶液沖洗離子交換樹脂,流速約為5~20mL/min,在沖洗過程中隨時對其進行檢測,使陰離子交換樹脂完全由Cl-型轉變為OH-,得到1-乙烯基-3-烷基咪唑離子液體;
然后,取1-乙烯基-3-烷基咪唑離子液體放入到溶解釜中,然后加入10~150份錸酸銨;加熱到70~80℃,反應10~20個小時;蒸發掉過量的水,加氯仿溶解,再加入引發劑;最后,放入真空烘箱中,80℃下真空干燥12~36h;最后得到液體即為1-乙烯基-3-烷基咪唑錸聚合離子液體。
本發明具有以下有益效果:
本發明方法,是一種成本低,產率高,不污染環境,可進行非均相催化高級烯烴環氧化反應,克服了催化劑在參與催化反應的同時自身結構變化的難題,與此同時,高級烯烴的環氧化物催化在國內就可進行,無需依賴國外進口,催化劑可回收和循環利用,有利于產品的分離純化的一種功能型錸聚合離子液體。即整個烯烴環氧化催化反應在特定的反應釜中進行,采用反應的溶劑,催化劑錸聚合離子液體、溶劑、氧化劑以及底物按照一定的先后順序加入到催化反應效果評價裝置中,其中錸聚合離子液體的量為加入總摩爾數的20%~70%,在恒定溫度50~100℃下攪拌反應,回流冷卻反應5~15h以后,從整個反應體系中,取出1~20份反應液,加入1~30份萃取劑充分振蕩萃取,用微量進樣器取上層溶液,用儀器對產物進行分析,用歸一法測得反應中得到產物的產率95%以上,反應循環使用5次以上,總轉化率均達到95%以上。
本發明采用錸聚合離子液體做催化劑,工藝簡單,提供一種成本低,產率高,不污染環境,可進行非均相催化高級烯烴環氧化反應,克服了催化劑在參與催化化學反應的同時自身結構變化的難題,催化劑可回收和循環利用,循環5次后轉化率仍在90%以上,且有利于產品的分離純化的一種功能型錸聚合離子液體。
附圖說明
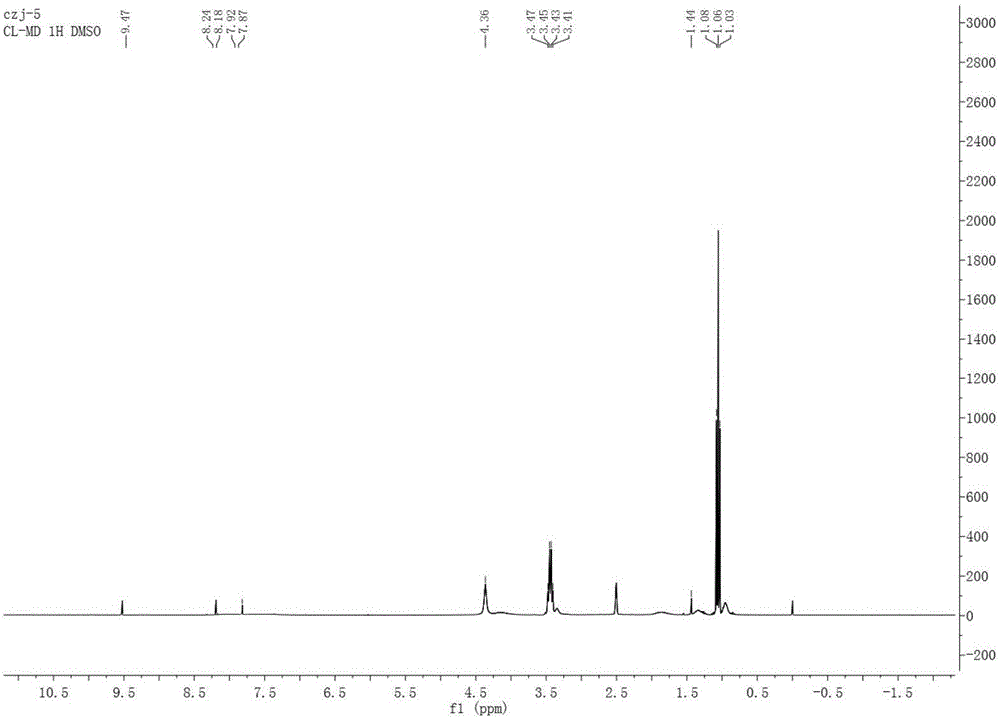
圖1為實施例1制備的1-乙烯基-3-丁基咪唑錸聚合離子液體P[BVIm][ReO4]1H-NMR譜圖。
圖2為實施例5歸一法測得錸聚合離子液體催化環辛烯氧化產率氣相譜圖。
具體實施方式
下面以具體的實施例進一步說明本發明的內容,但不應理解為對本發明的限制,在不背離本發明精神和實質的情況下,對本發明方法、步驟或是條件等所做修改或替換,均屬于本發明的范圍。
實施例1制備1-乙烯基-3-丁基咪唑錸聚合離子液體P[BVIm][ReO4]
以陽離子為1-乙烯基-3-丁基咪唑錸聚合離子液體(P[BVIm][ReO4])為例,其結構式如下:
其制備方法為:首先向N-乙烯基咪唑中,加入溴代烷烴,升溫至70~85℃反應5~15h后;冷卻至室溫,將產物放置于制冷機冷凍5~20h后取出,于85℃蒸發后,加入乙酸乙酯和乙腈的混合溶劑將結晶物溶解,再于85℃下蒸發重結晶,反復操作2-3次,再在70℃下真空干燥處理12~36h,得到紅棕色粘稠液體產品備用;準備一條陰離子交換樹脂柱,對陰離子交換樹脂除雜,把處理過的陰離子交換樹脂裝柱,裝入長100cm,內徑6cm離子交換柱中。然后用堿性溶液沖洗離子交換樹脂,流速約為5~20mL/min,在沖洗過程中隨時對其進行檢測,使陰離子交換樹脂完全由Cl-型轉變為OH-,得到1-乙烯基-3-丁基咪唑離子液體;
然后,取1-乙烯基-3-丁基咪唑離子液體放入到溶解釜中,然后加入10~150份錸酸銨;加熱到70~80℃,反應10~20個小時;蒸發掉過量的水,加氯仿溶解,再加入引發劑;最后,放入真空烘箱中,80℃下真空干燥12~36h;最后得到液體即為1-乙烯基-3-丁基咪唑錸聚合離子液體。
圖1為1-乙烯基-3-丁基咪唑錸聚合離子液體P[BVIm][ReO4]1H-NMR譜圖。由圖1可知HNMR(DMSO)δ=9.47、8.18、7.87附近有明顯的咪唑環上3個H的特征峰,δ=1.1附近是對應側鏈烷基上的H原子,δ=3~4附近為與咪唑環上氮原子相連的乙烯基聚合后的H原子,δ=2.5附近為溶劑帶來的雜峰。由于聚合物聚合時對原子的包裹作用,導致核磁共振氫譜譜線裂分效果不好,顯示在圖譜上則是在相應的化學位移處的譜線以“饅頭峰”出現。
實施例2、1-乙烯基-3-丁基咪唑錸聚合離子液體P[BVIm][ReO4]催化環戊烯環氧化的方法
其中,催化劑采用實施例1制備的1-乙烯基-3-丁基咪唑錸聚合離子液體P[BVIm][ReO4]。
整個烯烴環氧化催化反應在玻璃催化反應裝置中進行,將反應物:催化劑錸聚合離子液體P[BVIm][ReO4]、溶劑1,4-二氧六環、氧化劑過氧化脲以及底物環戊烯按照一定的先后順序加入到催化反應效果評價裝置中,其中錸聚合離子液體的量為加入總摩爾數的20%,氧化劑的摩爾數為環戊烯摩爾數的1.0倍,1,4-二氧六環與催化劑質量比為1:15,在恒定溫度100℃下磁力攪拌反應,回流冷卻反應3.5h。反應一定時間以后,從整個反應體系中,取出反應液加入萃取劑正己烷中,反應液與萃取劑正己烷的體積比1:2,充分振蕩萃取,用微量進樣器取上層溶液,取上層溶液的量占微量進樣器體積的50%,用氣相色譜儀對產物進行分析,得到單一產物,為該環戊烯的環氧化物,并以歸一法確認其定量指標,轉化率為95.4%。
在催化反應結束以后,加入正己烷劇烈攪拌2~5min以萃取體系中的底物和產物,再靜置待體系分層以后,取其上層正己烷溶液,如此反復5次以后,再加入正己烷萃取,取其上清液,再用氣相色譜儀進行檢測上清液,此時體系中底物和產物均已完全被萃取干凈。底物和產物萃取完成后,反應體系中只剩下溶劑離子液體、催化劑、氧化劑反應后留下的尿素以及少量的未反應的氧化劑。尿素和氧化劑都很容易溶于水中,因此用去離子水反復多次洗滌后,反應體系中只剩下催化劑,此時再將其80℃真空干燥24h即可,然后再將催化劑用于上述反應中。
實施例3、1-乙烯基-3-戊基咪唑錸聚合離子液體P[PVIm][ReO4]催化環己烯環氧化
整個烯烴環氧化催化反應在特定的反應釜中進行,將反應的溶劑,催化劑錸聚合離子液體P[PVIm][ReO4]、溶劑1,4-二氧六環、氧化劑過氧化脲以及底物烯烴按照一定的先后順序加入到催化反應效果評價裝置中,其中錸聚合離子液體的量為加入總摩爾數的70%,氧化劑的摩爾數為烯烴摩爾數的2.0倍,溶劑與催化劑質量比為1:10,在恒定溫度60℃下磁力攪拌反應,回流冷卻反應3h。反應一定時間以后,從整個反應體系中,取出反應液與萃取劑正己烷,反應液與萃取劑正己烷的體積比1:3,充分振蕩萃取,用微量進樣器取上層溶液,取上層溶液的量占微量進樣器體積的65%,用氣相色譜儀對產物進行分析,得到單一產物,為該烯烴的環氧化物,并以歸一法確認其定量指標,轉化率為96.8%,反應體系經簡單處理或不處理進行循環實驗,待循環5次后轉化率仍在95%以上。測得反應中得到產物的產率為95%。
實施例4、1-乙烯基-3-己基咪唑錸聚合離子液體P[HVIm][ReO4]催化環庚烯環氧化
整個烯烴環氧化催化反應在特定的反應釜中進行,將反應的溶劑,催化劑錸聚合離子液體P[HVIm][ReO4]、溶劑1,4-二氧六環、氧化劑過氧化脲以及底物烯烴按照一定的先后順序加入到催化反應效果評價裝置中,其中錸聚合離子液體的量為加入總摩爾數的50%,氧化劑的摩爾數為烯烴摩爾數的2.5倍,溶劑與催化劑質量比為1:20,在恒定溫度50℃下磁力攪拌反應,回流冷卻反應5h。反應一定時間以后,從整個反應體系中,取出反應液與萃取劑正己烷,反應液與萃取劑正己烷的體積比1:5,充分振蕩萃取,用微量進樣器取上層溶液,取上層溶液的量占微量進樣器體積的70%,用氣相色譜儀對產物進行分析,得到單一產物,為該烯烴的環氧化物,并以歸一法確認其定量指標,轉化率為97%,反應體系經簡單處理或不處理進行循環實驗,待循環5次后轉化率仍在95%以上,測得反應中得到產物的產率為95.4%。
實施例5、1-乙烯基-3-乙基咪唑錸聚合離子液體P[EVIm][ReO4]催化環辛烯環氧化
整個烯烴環氧化催化反應在特定的反應釜中進行,將反應的溶劑,催化劑錸聚合離子液體P[EVIm][ReO4]、溶劑1,4-二氧六環、氧化劑過氧化脲以及底物烯烴按照一定的先后順序加入到催化反應效果評價裝置中,其中錸聚合離子液體的量為加入總摩爾數的40%,氧化劑的摩爾數為烯烴摩爾數的3.0倍,溶劑與催化劑質量比為1:25,在恒定溫度80℃下磁力攪拌反應,回流冷卻反應10h。反應一定時間以后,從整個反應體系中,取出反應液與萃取劑正己烷,反應液與萃取劑正己烷的體積比1:4,充分振蕩萃取,用微量進樣器取上層溶液,取上層溶液的量占微量進樣器體積的80%~90%,用氣相色譜儀對產物進行分析,得到單一產物,為該烯烴的環氧化物,并以歸一法確認其定量指標,轉化率為97.2%,反應體系經簡單處理或不處理進行循環實驗,待循環5次后轉化率仍在95%以上,測得反應中得到產物的產率為96%。
圖2為本實施例歸化一法測得錸聚合離子液體催化環辛烯氧化產率氣相譜圖。由圖2可知,環辛烯完全轉化為環氧化物,無其他副產物生成。
- 還沒有人留言評論。精彩留言會獲得點贊!
- 一種錸聚合離子液體催化烯烴環氧化的方法與制造工藝
- 一種制備高純度烯烴的催化劑的制備方法與制造工藝
- 稀土催化劑組合物及其制備方法和共軛二烯烴的聚合方法與制造工藝
- 一種催化劑的制備方法和制得的催化劑及其應用與制造工藝
- 一種用于烯烴聚合的催化劑體系及其應用的制造方法與工藝
- 用于烯烴聚合的催化劑組分及其制備方法和應用和用于烯烴聚合的催化劑及其應用與制造工藝
- 用于烯烴聚合的催化劑組分及其制備方法和應用和用于烯烴聚合的催化劑及其應用與制造工藝
- 用于烯烴聚合的催化劑組分、催化劑及其應用的制造方法與工藝
- 用于烯烴聚合的催化劑組分及其制備方法和應用和用于烯烴聚合的催化劑及其應用與制造工藝
- 用于烯烴聚合的催化劑組分及其制備方法和應用和用于烯烴聚合的催化劑及其應用與制造工藝