以磷改性氧化鋁為基質原位合成SAPO?11@γ?Al2O3復合載體材料的方法與流程
本發明涉及石油化工技術領域,具體的說,本發明涉及一種以磷改性氧化鋁為基質原位合成SAPO-11@γ-Al2O3復合載體材料的方法。
背景技術:
SAPO-11分子篩是1984年美國UOP公司首先研發(US4440871)的一種磷酸硅鋁分子篩。SAPO-n是由Si原子取代AlPO4-n骨架中的P或Al原子后形成的由AlO4、PO4、SiO4四面體構成分子篩骨架結構。作為SAPO-n分子篩家族的一員,SAPO-11分子篩以其獨特的一維十元環直型孔道結構及可調變的酸性,在正構烷烴的異構化反應中表現出較高的活性和選擇性,可以用于烷烴異構化反應催化劑的酸性組分。
我們的研究發現,催化材料在工程催化劑中的有效比表面積取決于兩個方面:一是材料的本征性質及其合成方法;二是催化劑的工程制備方法,不論采用何種方法制取的分子篩材料均為粉體材料,在工業應用中通常要對粉體催化劑進行成型處理,以克服其易流失、傳質阻力大等缺點。
原位晶化技術是將分子篩組分原位生長在某種基質上的一種制備方法。原位晶化技術將沸石活性組分的晶化合成過程與催化劑載體的制備過程有機地結合在一起,使分子篩晶粒在基質表面原位生長并附著,從而得到一類分子篩@基質復合材料。
然而基質材料的表面特性對分子篩原位晶化也具有重要影響,不可溶解的基質可以提供物理支撐,承載SAPO-11分子篩的生長,所生成的SAPO-11覆蓋基質表面;可溶解的基質(如黏土和氧化鋁)由于酸溶液的抽提作用而溶解,借以改變基質表面的液相組成,基質表面特性在SAPO-11分子篩原位晶化過程中具有一定的結構導向作用。γ-Al2O3由致密的氧化鋁相構成的微晶堆積而成,微晶間不規則的空位形成了孔徑分布寬的三維交叉孔道。γ-Al2O3基質中的活性鋁物質在SAPO-11分子篩合成體系中的弱酸作用下不斷溶解于溶液之中,活性鋁溶出后留出的基質表面繼續與母液中的活性磷、鋁、硅等組分作用,成為SAPO-11分子篩生長的“活性位”和“生長點”,而溶液中的硅源、鋁源及模板劑等原料也持續向基質經酸液侵蝕溶出的表面孔道擴散,基質與溶液主體截然不同的組成使γ-Al2O3表面的化學組成并不一致。因此,在合成體系中,基質與溶液組成上的差異使其交界面存在一定的濃度梯度,該濃度差一方面使反應原料在基質-溶液的交界面上發生持續的物質傳遞,另一方面使得SAPO-11在γ-Al2O3基質上的原位晶化生長處于動態的物質傳遞平衡過程之中。但γ-Al2O3基質中活性鋁的過度溶解會造成基質機械強度下降,且母液中過高的Al含量對合成體系配比要求較為嚴格的SAPO-11分子篩的合成是不利的。因此,基質孔道形態的選擇及基質活性鋁過度溶解的避免,是成功合成SAPO-11@γ-Al2O3復合載體材料的關鍵步驟之一。
CN101069854A公開了一種改性MCM-41/氧化鋁復合載體材料的制備方法,通過將氧化鋁在可溶性氟化物溶液中進行浸漬改性,水熱合成了MCM-41/氧化鋁復合材料,提高了MCM-41的結構穩定性,調節復合材料的表面酸堿度,使其更適合于作為柴油加氫脫硫等加氫處理催化劑的載體。
文獻Langmuir,2009,25:2271-2277中以異丙醇鋁為鋁源,磷酸為磷源,正硅酸乙酯為硅源,三乙胺為模板劑,通過水熱合成法在殼聚糖覆蓋的α-Al2O3微球上原位生長SAPO-5分子篩。其所合成的復合材料中的SAPO-5含量較高,但是為了避免α-Al2O3微球在合成體系中的溶解,作者使用殼聚糖對氧化鋁微球進行表面浸涂處理,其改性過程過于復雜,不利于工業批量生產。
文獻Applied Catalysis A:General,2008,341:112-118以擬薄水鋁石為鋁源,硅溶膠為硅源,三乙胺為模板劑,磷酸為磷源,通過水熱合成法在α-Al2O3微球上原位生長SAPO-34分子篩,但其所合成的復合材料中SAPO-34的生長量較低。
梯級孔分子篩兼具有序介孔材料和微孔分子篩的優點,可以提高雙支鏈及多支鏈異構烷烴的選擇性。目前制備梯級孔硅鋁分子篩主要有三種方法:基于脫硅或脫鋁的后處理方法、基于硬(軟)模板的直接合成法和基于納米晶組裝的方法。由于后處理方法所形成的介孔尺寸不均勻、不可控,且骨架硅鋁的過度脫除影響分子篩的結晶度與穩定性,尤為重要的是,SAPO分子篩承受水熱處理的能力不及硅鋁分子篩,故此方法不適用。硬模板法所采用的模板劑價格便宜,但所形成的介孔連通性差,且硬模板劑親水性弱,在合成過程中易發生相分離,難以應用于工業放大。
納米晶組裝技術有別于常規的介孔材料合成方法,它以納米晶/簇(nanoclusters)或晶種(seeds)為結構單元取代無定形硅鋁物種作為合成原料,通過表面活性劑的模板導向作用,形成孔壁含有微孔沸石結構單元的介孔材料。
文獻Journal of the American Chemical Society,2000,122:8791-8792首先制備出了含有Y型沸石的納米晶,通過CTABr進行自組裝獲得了具有六方排列的介孔分子篩MSU-S,該材料具有比“超穩”介孔硅鋁分子篩更高的穩定性及比Al-MCM-41更優的催化性能。
文獻Journal of the American Chemical Society,2003b,125:2376-2377采用Beta沸石的納米晶、以玉米淀粉為模板劑,組裝合成了具有9~20nm孔道的非規則介孔材料,其水熱穩定性顯著提高。
文獻Journal of Catalysis,2007,251:69-79首先在堿性環境中將CTABr預先錨定于高嶺土表面形成有序的模板膠束,再將Y沸石納米晶在模板膠束的作用下原位組裝合成介孔分子篩,得到MY/kaolin復合物材料,該復合物催化劑對重油的裂化反應表現出了優異的催化性能。
由于可以選擇不同的沸石納米晶以及不同的表面活性劑模板膠束進行組裝,這更增加了合成過程與合成產品的靈活性和多樣性,從而降低催化劑制備的成本。盡管納米晶組裝方法已在硅鋁分子篩的合成中得到了廣泛應用,但目前尚無通過納米晶組裝法合成梯級孔磷酸硅鋁分子篩的報道。因此,基于軟模板的納米晶組裝方法成為構筑具有梯級孔結構SAPO-11@γ-Al2O3復合載體材料值得探索的重要方法。
目前有關SAPO-11分子篩的合成研究大多致力于改善SAPO-11分子篩的酸性和孔結構,而對其原位合成技術的研究較少。為有效解決上述問題,本發明提供了一種以磷改性氧化鋁為基質原位合成SAPO-11@γ-Al2O3復合載體材料的方法,該方法采用獨特的磷改性方法避免了氧化鋁基質在合成體系中的溶解,并將納米晶的組裝、原位合成與催化劑的制備過程有機結合起來,在所獲得SAPO-11@γ-Al2O3復合載體材料中,SAPO-11分子篩暴露于載體的外表面,從而顯著增加了分子篩的有效比表面積,提高了分子篩的利用率,簡化了催化劑載體的制備工藝,降低了催化劑載體的制備成本。
技術實現要素:
本發明的一個目的在于提供一種以磷改性氧化鋁為基質原位合成SAPO-11@γ-Al2O3復合載體材料的方法;
本發明的另一目的在于提供所述的方法制備得到的SAPO-11@γ-Al2O3復合載體材料。
為達上述目的,一方面,本發明提供了以一種磷改性氧化鋁為基質原位合成SAPO-11@γ-Al2O3復合載體材料的方法,其中,所述方法包括如下步驟:
(1)氧化鋁基質改性:使用磷酸水溶液對氧化鋁基質進行水熱改性得到改性的氧化鋁基質;
(2)SAPO-11分子篩前驅體溶液的制備:將鋁源、磷酸、有機模板劑、硅源和水混合均勻,然后預晶化;
(3)凝膠混合物的制備:將步驟(2)得到的產物在攪拌下加入到表面活性劑溶液中,然后老化處理,得到凝膠混合物;
(4)復合載體材料的制備:將步驟(1)得到的改性的氧化鋁基質和步驟(3)得到的凝膠混合物混合后晶化,然后經過處理后焙燒得到所述的SAPO-11@γ-Al2O3復合載體材料。
根據本發明一些具體實施方案,其中,步驟(1)所述水熱改性的溫度是80-180℃,改性的時間是4-48h。
根據本發明一些具體實施方案,其中,步驟(1)的磷酸水溶液與氧化鋁基質的質量比為5:1-10:1。
根據本發明一些具體實施方案,其中,步驟(1)的磷酸水溶液中磷酸與水的質量比為1:30-1:20。
步驟(1)的改性可以在本領域常用的晶化釜中進行,根據本發明一些具體實施方案,步驟(1)的改性是在襯有聚四氟乙烯的晶化釜中進行的。
根據本發明一些具體實施方案,其中,步驟(2)中磷源、鋁源和硅源分別以P2O5、Al2O3、SiO2計,磷源、鋁源、硅源、有機模板劑及去離子水的摩爾比為(1.5~2):1:(0.1~0.2):(1~3):(150~300)。
根據本發明一些具體實施方案,其中,步驟(2)中所述預晶化的溫度為100-160℃,時間為4-24h。
根據本發明一些具體實施方案,其中,步驟(2)是將鋁源、磷源、有機模板劑、硅源加入水中后,在20-80℃下攪拌8-24h,然后預晶化。
根據本發明一些具體實施方案,其中,步驟(2)是將鋁源、磷源、有機模板劑和硅源按順序依次加入水中。
根據本發明一些具體實施方案,其中,步驟(2)每加入一種成分后,都要攪拌4-6h。
也就是說,步驟(2)每加入一種成分后,都要攪拌4-6h,然后再加入另一成分。
步驟(2)的預晶化可以在本領域常用的晶化釜中進行,根據本發明一些具體實施方案,步驟(2)的預晶化是在襯有聚四氟乙烯的晶化釜中進行的。
根據本發明一些具體實施方案,其中,步驟(3)中老化溫度為40-60℃,時間為8-24h。
根據本發明一些具體實施方案,其中,步驟(3)的凝膠混合物中磷源、鋁源、硅源分別以P2O5、Al2O3、SiO2計,與有機模板劑、表面活性劑及去離子水的摩爾比為(1.5~2):1:(0.1~0.2):(1~3):(0.01~0.03):(150~300)。
根據本發明一些具體實施方案,其中,步驟(4)中所述凝膠混合物與改性的氧化鋁基質的質量比為5:1-20:1。
根據本發明一些具體實施方案,其中,步驟(4)的晶化條件為:在自生壓力下,150-200℃下晶化24-72h。
根據本發明一些具體實施方案,其中,步驟(4)晶化后,經過過濾、洗滌、干燥、然后焙燒得到所述的SAPO-11@γ-Al2O3復合載體材料。
根據本發明一些具體實施方案,其中,步驟(4)晶化后,經過過濾、洗滌、干燥、然后在500-700℃下焙燒5-7h得到所述的SAPO-11@γ-Al2O3復合載體材料。
根據本發明一些具體實施方案,其中,所述氧化鋁基質為工業成型氧化鋁。
其中可以理解的是,所述的工業成型氧化鋁是指將工業上以氧化鋁粉為原料經過成型加工后得到的成型氧化鋁球或氧化鋁條等;本發明將這種以氧化鋁粉為原料經過成型加工后得到的成型氧化鋁球或氧化鋁條等直接用作氧化鋁基質,不需要進行任何進一步的加工。
根據本發明一些具體實施方案,其中,所述氧化鋁基質選自氧化鋁球、三葉草狀氧化鋁條、柱狀氧化鋁條、蜂窩狀氧化鋁、齒球狀氧化鋁、和環狀氧化鋁中的一種或幾種。
根據本發明一些具體實施方案,其中,所述氧化鋁球直徑為2-3mm。
根據本發明一些具體實施方案,其中,所述三葉草狀氧化鋁條直徑為1-3mm。
根據本發明一些具體實施方案,其中,所述柱狀氧化鋁條直徑為1-3mm。
根據本發明一些具體實施方案,其中,所述鋁源選自擬薄水鋁石、異丙醇鋁、硫酸鋁、硝酸鋁和鋁溶膠中的一種或幾種;
根據本發明一些具體實施方案,其中,所述磷源選自磷酸;
根據本發明一些具體實施方案,其中,所述有機模板劑選自二乙胺、二正丙胺、二異丙胺、二正丁胺和四丙基溴化銨中的一種或幾種;
根據本發明一些具體實施方案,其中,所述硅源選自硅溶膠、正硅酸乙酯和四丙氧基硅烷中的一種或幾種。
根據本發明一些具體實施方案,其中,所述表面活性劑選自十烷基三甲基溴化銨、十二烷基三甲基溴化銨、十四烷基三甲基溴化銨和十六烷基三甲基溴化銨(CTABr)中的一種或幾種。
另一方面,本發明還提供了所述的方法制備得到的SAPO-11@γ-Al2O3復合載體材料。
根據本發明一些具體實施方案,其中,所述復合材料中SAPO-11的相對結晶度為33%-42%(利用粉末X射線衍射(XRD)確定);SAPO-11相的含量為32wt.%-41wt.%(由XRD的“內標法”確定)。
復合載體材料中SAPO-11含量與SAPO-11相對結晶度的對照關系如圖1所示。同時該復合材料剖面的掃描電鏡(SEM)照片顯示出兩個明顯不同的結構區域,分別是由氧化鋁基質組成的內核與外部覆蓋的SAPO-11分子篩層,表明該復合物具有典型的“核-殼”型結構,直接反映出該復合物中SAPO-11相和氧化鋁基質之間是通過化學鍵的作用方式相互連接的,而不是簡單的機械混合。
綜上所述,本發明提供了一種以磷改性氧化鋁為基質原位合成的SAPO-11@γ-Al2O3復合載體材料的方法。本發明的方案具有如下優點:直接在磷改性后的工業成型氧化鋁基質上進行SAPO-11分子篩的原位生長,使分子篩全部暴露在催化劑載體的外表面,有利于反應物與活性中心的充分接觸,提高了分子篩材料的利用率,簡化了催化劑載體的制備過程。
附圖說明
圖1為機械混合物中SAPO-11含量與SAPO-11相對結晶度的對照關系圖。
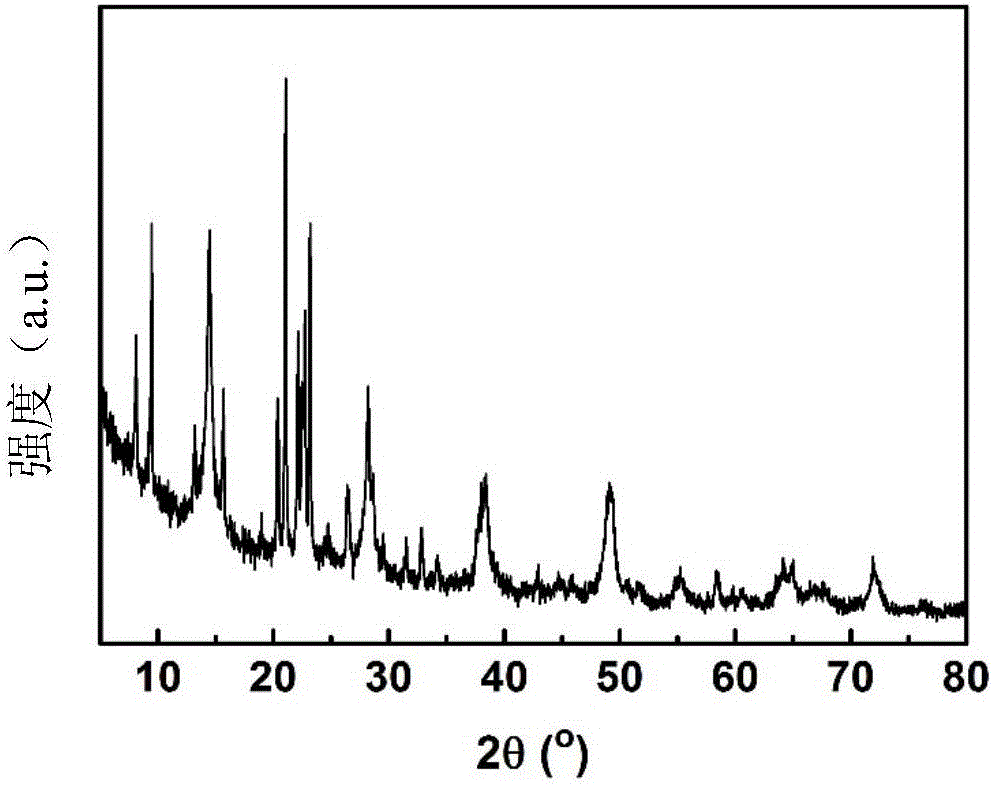
圖2為本發明實施例1所得復合材料的XRD譜圖。
圖3為本發明實施例1所得復合材料放大30倍的SEM照片。
圖4為本發明實施例1所得復合材料放大130倍的SEM照片。
圖5-圖14為本發明實施例2-例11所得復合材料的XRD譜圖。
具體實施方式
以下通過具體實施例詳細說明本發明的實施過程和產生的有益效果,旨在幫助閱讀者更好地理解本發明的實質和特點,不作為對本案可實施范圍的限定。
實施例1
(1)氧化鋁基質的改性:將2.4g磷酸加入到60g去離子水中得到磷酸水溶液,將該水溶液與10g氧化鋁球混合后轉移至內襯聚四氟乙烯的晶化釜中,在150℃下水熱改性24h,產物經過濾、洗滌,在120℃干燥4h后,得到磷改性的氧化鋁基質。
(2)SAPO-11分子篩納米晶前驅體溶液的制備:在80℃下將8g異丙醇鋁加入到60g去離子水中,攪拌2h;滴加7g磷酸,攪拌2h;逐滴加入4.95g二異丙胺,攪拌2h;加入1g四丙氧基硅烷,攪拌2h后將該混合物置于內襯聚四氟乙烯的晶化釜中,在160℃下預晶化12h,得到SAPO-11納米晶前驅體溶液。
(3)凝膠混合物的制備:將0.2g CTABr溶于20g去離子水中,攪拌2h;將(2)所獲得的前驅體溶液于攪拌狀態下滴加入該表面活性劑溶液中,40℃條件下老化24h,得到凝膠混合物。
(4)復合載體材料的制備:將上述凝膠混合物80g與5g改性氧化鋁基質置于晶化釜中,在185℃、自生壓力下晶化48h后,將晶化產物取出,經去離子水洗滌、過濾、120℃干燥4h、600℃焙燒6h后,得到SAPO-11@γ-Al2O3復合材料A。
經XRD表征,產物中SAPO-11分子篩的相對結晶度為42%。其XRD譜圖見圖2,SEM照片見圖3、圖4。
實施例2
按照實施例1的制備方法,制備SAPO-11@γ-Al2O3復合材料B。與實施例1不同的是前驅體溶液配制過程的投料順序。
(1)氧化鋁基質的改性:同實施例1。
(2)SAPO-11分子篩納米晶前驅體溶液的制備:將7g磷酸加入到60g去離子水中,在80℃攪拌2h;加入8g異丙醇鋁,攪拌6h;逐滴加入4.95g二異丙胺,攪拌4h;加入1g四丙氧基硅烷,攪拌4h后將該混合物置于內襯聚四氟乙烯中的晶化釜中,在160℃下預晶化12h,得到SAPO-11納米晶前驅體溶液。
(3)凝膠混合物的制備:同實施例1。
(4)復合載體材料的制備:同實施例1。
產物中SAPO-11分子篩的相對結晶度為40%。其XRD譜圖見圖5。
實施例3
按照實施例1的制備方法,制備SAPO-11@γ-Al2O3復合材料C。與實施例1不同的是采用了不同的硅源。
(1)氧化鋁基質的改性:同實施例1。
(2)SAPO-11分子篩納米晶前驅體溶液的制備:在80℃下將8g異丙醇鋁加入到60g去離子水中,攪拌4h;滴加7g磷酸,攪拌2h;逐滴加入4.95g二異丙胺;加入0.79g正硅酸乙酯,攪拌4h后將該混合物置于內襯聚四氟乙烯中的晶化釜中,在160℃下預晶化12h,得到SAPO-11納米晶前驅體溶液。
(3)凝膠混合物的制備:同實施例1。
(4)復合載體材料的制備:同實施例1。
產物中SAPO-11分子篩的相對結晶度為39%。其XRD譜圖見圖6。
實施例4
按照實施例1的制備方法,制備SAPO-11@γ-Al2O3復合材料D。與實施例1不同的是采用了不同的鋁源。
(1)氧化鋁基質的改性:同實施例1。
(2)SAPO-11分子篩納米晶前驅體溶液的制備:在80℃下將2g擬薄水鋁石加入到60g去離子水中,攪拌5h;滴加7g磷酸,攪拌3h;逐滴加入4.95g二異丙胺,攪拌4h;加入1g四丙氧基硅烷,攪拌4h將該混合物置于內襯聚四氟乙烯中的晶化釜中,在160℃下預晶化12h,得到SAPO-11納米晶前驅體溶液。
(3)凝膠混合物的制備:同實施例1。
(4)復合載體材料的制備:同實施例1。
產物中SAPO-11分子篩的相對結晶度為33%。其XRD譜圖見圖7。
實施例5
按照實施例1的制備方法,制備SAPO-11@γ-Al2O3復合材料E。與實施例1不同的是采用了不同的表面活性劑。
(1)氧化鋁基質的改性:同實施例1。
(2)SAPO-11分子篩納米晶前驅體溶液的制備:同實施例1。
(3)凝膠混合物的制備:將0.17g十二烷基三甲基溴化銨溶于20g去離子水中,攪拌2h;將(2)所獲得的前驅體溶液于攪拌狀態下滴加入該表面活性劑溶液中,40℃條件下老化24h,得到凝膠混合物。
(4)復合載體材料的制備:同實施例1。
產物中SAPO-11分子篩的相對結晶度為39%。其XRD譜圖見圖8。
實施例6
按照實施例1的制備方法,制備SAPO-11@γ-Al2O3復合材料F。與實施例1不同的是采用了不同的有機模板劑。
(1)氧化鋁基質的改性:同實施例1。
(2)SAPO-11分子篩納米晶前驅體溶液的制備:在80℃下將8g異丙醇鋁加入到60g去離子水中,攪拌2h;滴加7g磷酸,攪拌2h;逐滴加入4.95g二正丙胺,攪拌2h;加入1g四丙氧基硅烷,攪拌2h后將該混合物置于內襯聚四氟乙烯中的晶化釜中,在160℃下預晶化12h,得到SAPO-11納米晶前驅體溶液。
(3)凝膠混合物的制備:同實施例1。
(4)復合載體材料的制備:同實施例1。
產物中SAPO-11分子篩的相對結晶度為39%。其XRD譜圖見圖9。
實施例7
按照實施例1的制備方法,制備SAPO-11@γ-Al2O3復合材料G。與實施例1不同的是前驅體溶液制備過程的攪拌溫度。
(1)氧化鋁基質的改性:同實施例1。
(2)SAPO-11分子篩納米晶前驅體溶液的制備:在40℃將8g異丙醇鋁加入到60g去離子水中,攪拌2h;滴加7g磷酸,攪拌2h;逐滴加入4.95g二異丙胺,攪拌2h;加入1g四丙氧基硅烷,攪拌2h后將該混合物置于內襯聚四氟乙烯中的晶化釜中,在160℃下預晶化12h,得到SAPO-11納米晶前驅體溶液。
(3)凝膠混合物的制備:同實施例1。
(4)復合載體材料的制備:同實施例1。
產物中SAPO-11分子篩的相對結晶度為36%。其XRD譜圖見圖10。
實施例8
按照實施例1的制備方法,制備SAPO-11@γ-Al2O3復合材料H。與實施例1不同的是使用不同形貌的氧化鋁基質。
(1)氧化鋁基質的改性:將4g磷酸加入到100g去離子水得到磷酸水溶液,將該水溶液與10g長度為2~4mm、直徑為1~2mm的三葉草狀氧化鋁條狀基質混合后轉移至內襯聚四氟乙烯中的晶化釜中,在150℃下水熱改性24h,產物經過濾、洗滌,在120℃干燥4h后,得到改性氧化鋁基質。
(2)SAPO-11分子篩納米晶前驅體溶液的制備:同實施例1。
(3)凝膠混合物的制備:同實施例1。
(4)復合載體材料的制備:同實施例1。
產物中SAPO-11分子篩的相對結晶度為36%。其XRD譜圖見圖11。
實施例9
按照實施例1的制備方法,制備SAPO-11@γ-Al2O3復合材料I。與實施例1不同的是晶化溫度。
(1)氧化鋁基質的改性:同實施例1。
(2)SAPO-11分子篩納米晶前驅體溶液的制備:同實施例1。
(3)凝膠混合物的制備:同實施例1。
(4)復合載體材料的制備:將上述凝膠混合物80g與5g改性氧化鋁基質置于晶化釜中,在200℃、自生壓力下晶化48h后,將晶化產物取出,經去離子水洗滌、過濾、120℃干燥4h、600℃焙燒6h后,得到SAPO-11@γ-Al2O3復合材料I。
產物中SAPO-11分子篩的相對結晶度為35%。其XRD譜圖見圖12。
實施例10
按照實施例1的制備方法,制備SAPO-11@γ-Al2O3復合材料J。與實施例1不同的是晶化時間。
(1)氧化鋁基質的改性:同實施例1。
(2)SAPO-11分子篩納米晶前驅體溶液的制備:同實施例1。
(3)凝膠混合物的制備:同實施例1。
(4)復合載體材料的制備:將上述凝膠混合物80g與5g改性氧化鋁基質置于晶化釜中,在185℃、自生壓力下晶化24h,將晶化產物取出,經去離子水洗滌、過濾、120℃干燥4h、600℃焙燒6h后,得到SAPO-11@γ-Al2O3復合材料J。
產物中SAPO-11分子篩的相對結晶度為34%。其XRD譜圖見圖13。
實施例11
按照實施例1的制備方法,制備SAPO-11@γ-Al2O3復合材料K。與實施例1不同的是前驅體溶液與改性氧化鋁基質的投料比。
(1)氧化鋁基質的改性:同實施例1。
(2)SAPO-11分子篩納米晶前驅體溶液的制備:同實施例1。
(3)凝膠混合物的制備:同實施例1。
(4)復合載體材料的制備:將上述凝膠混合物60g與5g改性氧化鋁基質置于晶化釜中,在185℃、自生壓力下晶化48h后,將晶化產物取出,經去離子水洗滌、過濾、120℃干燥4h、600℃焙燒6h后,得到SAPO-11@γ-Al2O3復合材料K。
產物中SAPO-11分子篩的相對結晶度為34%。其XRD譜圖見圖14。
實施例1-11催化性能評價
以實施例1-11所制得的SAPO-11@γ-Al2O3復合材料為載體,采用等體積共浸漬法,擔載1.0wt.%的NiO和3.0wt.%的MoO3(以催化劑重量計),得到Ni-Mo/SAPO-11@γ-Al2O3催化劑。
以格爾木全餾分催化裂化(FCC)汽油為原料油,考察并評價所制得的Ni-Mo/SAPO-11@γ-Al2O3催化劑的臨氫異構反應性能。評價條件如下:反應壓力1.5MPa,反應溫度320℃,氫油體積比400:1,重時空速1.5h-1,原料組成及裝置運轉20h后的反應結果如表1。
表1 Ni-Mo/SAPO-11@γ-Al2O3催化劑上FCC汽油異構化反應結果
通過反應結果可以發現,在臨氫異構化反應中,用本發明所合成的復合載體材料所制備的催化劑可以有效的降低FCC汽油中的烯烴含量,提升異構烷烴的含量,從而維持產品辛烷值的不降低,同時能夠一定程度的降低油品的硫含量。這說明以SAPO-11@γ-Al2O3復合材料為載體,擔載非貴金屬所制得的Ni-Mo/SAPO-11@γ-Al2O3催化劑具有較優的異構化性能和降烯烴性能,并維持產品辛烷值,有較佳的研究和應用前景。
- 還沒有人留言評論。精彩留言會獲得點贊!