一種三維網狀結構MoO2納米材料及其制備和應用的制作方法
本發明屬于高能電池材料技術領域,具體涉及一種高性能具有三維網狀結構MoO2負極材料及其制備方法和應用。
背景技術:
鋰離子電池憑借較高的能量密度及顯著的性價比,目前被廣泛用于3C電子產品,電動汽車及規模儲能領域。近年來新能源汽車的迅速發展刺激了鋰離子電池的需求量急劇上漲,同時市場對鋰離子電池的能量密度提出了更高的要求。傳統的鋰離子電池石墨負極材料理論比容量較低,難于滿足下一代高能量密度鋰離子電池的要求,因此發展下一代高容量鋰離子電池負極材料顯得尤為迫切。
MoO2生產成本低廉,制備方法簡單,該材料具有較高的振實密度和能與金屬相媲美的導電性,作為鋰離子電池負極材料具有非常高的理論比容量(838mAh g-1),應用前景廣闊。另外我國鉬資源豐富,開發高容量的鉬系化合物作為新型鋰離子電池嵌鋰材料,對優化我國鉬資源利用和促進經濟發展都具有重要意義。目前MoO2在鋰離子電池應用中仍面臨以下問題,首先,MoO2在充放電過程中初步鋰化形成低導電性的Li0.98MoO2,阻止了MoO2材料的進一步鋰化,難于發揮其高儲鋰容量特性(Hu,X;Zhang,W;Liu,X et al.Chem Soc Rev 2015,44,2376-2404)。其次在鋰化過程中晶胞會有很大的體積膨脹,因此沒有經過改性的MoO2大顆粒材料比容量低且循環性能差。目前改善方法主要包括制備納米尺度MoO2材料,調控納米形貌,構筑多維結構來提高鋰離子和電子的遷移速率和抗體積膨脹的能力。如MoO2納米粒子/石墨烯復合材料(Petnikota,S;Teo,K;Chen,L et al.ACS Appl Mater Interfaces 2016,8,10884-10896)、介孔MoO2(Liu,D;Zheng,F;Su,J et al.J Mater Chem A.2016,4,12434-12441)、空心MoO2納米球(Tang,S;Ouyang,B;Yang,L et al.RSC Advances 2015,5,50705-50710)和MoO2納米片(Zhang,H;Zeng,L;Wu X et al.J Alloy Compd 2013,580,358-362)。雖然納米化能提高了MoO2的循環性能,但是簡單的低維MoO2納米材料由于其高的比表面積在循環過程中面臨著活性物質團聚的問題,使得MoO2在長循環過程中容量衰減較快。大尺寸的三維納米材料相比簡單的低維納米材料具有更好的結構穩定性,特別是三維納米網狀結構材料,綜合了納米材料高的比表面積和微米尺寸材料結構穩定抗團聚的優勢,在循環過程中能保持更好的電化學穩定性。然而據我們所知,目前仍然沒有合成三維網狀結構MoO2納米材料的相關報道。
本發明涉及了一種三維網狀結構MoO2的合成方法,通過一個攪拌的油水兩相水熱體系合成了網狀的納米三氧化鉬(MoO3)前驅體,然后通過煅燒后處理得到了三維網狀結構MoO2。相比于簡單的低維MoO2納米材料(納米粒子、納米棒和納米片等),該三維網狀結構MoO2的循環穩定性能和倍率性能都得到了顯著提高。
技術實現要素:
本發明的目的在于提供一種三維網狀結構的MoO2納米材料及其制備方法和應用。該法流程簡單、操作方便、生產成本低、適合規模化生產。用該方法制備的MoO2材料結構新穎,作為鋰離子電池負極材料具有優異的電化學性能。
一種三維網狀結構MoO2納米材料的制備方法,包括以下步驟:
將鉬源化合物溶于一定量水中,調節溶液pH并加入絡合劑,最后加入有機溶劑,經攪拌水熱反應得到前驅體,冷卻至室溫,經離心,洗滌,干燥得到前驅體粉末,之后將前驅體在惰性或還原性氣氛下燒結處理,得到三維網狀結構的MoO2納米材料。
所述的制備方法,所述的鉬源化合物包絡鉬酸,鉬酸銨,鉬酸鈉中的一種或幾種,鉬源化合物的濃度范圍在0.01~1mol/L。
所述的制備方法,所調節的pH范圍在0~6。
所述的制備方法,所用的絡合劑包括檸檬酸鈉,檸檬酸,抗壞血酸,苯甲酸中的一種或幾種,絡合劑的添加量為鉬源化合物的物質量的0.5~2倍。
所述的制備方法,所述的有機溶劑包括氯仿、甲苯、二甲苯、油酸中的一種或幾種,有機溶劑的添加量為鉬源化合物水溶液體積的1~3倍。
所述的制備方法,水熱反應溫度為150~200℃,時間3~48小時。
所述的制備方法,攪拌輔助水熱反應,攪拌轉速為300~600r/min。
所述的制備方法,所述的升溫燒結處理的溫度為200~800℃;燒結處理0.5~6小時。
所述的制備方法,所述的惰性或還原性氣體為:純Ar,N2氣,H2與Ar的混合氣體,或者CO與CO2的混合氣體。H2與Ar的混合氣體中H2體積分數為3~20%;CO與CO2的混合氣體中CO的體積分數為5~20%。
一種三維網狀結構MoO2納米材料,是由上述的方法制備而成的。
所述的三維網狀結構MoO2納米材料的應用,用于制備鋰離子電池負極材料。
本發明的原理:
鉬酸根離子(MoO42-)在不同濃度的酸性溶液里會脫水縮合形成不同形態的多鉬酸根離子([HxMo2O6]x+),從而[HxMo2O6]x+的荷電狀態及自組裝性質都會受到很大影響。此外[HxMo2O6]x+隨著反應釜內溫度和壓力的升高會進一步脫水縮合形成MoO3。因為有機溶劑如:甲苯與水不互溶,在劇烈的攪拌情況下甲苯以小液滴的形式分散在水相中并提供很高的甲苯/水兩相界面。由于界面張力的作用,特定的[HxMo2O6]x+被吸附到甲苯/水界面處發生自組裝生成MoO3納米線。同時在攪拌作用下MoO3納米線受力不均而彎曲相互纏繞交織生成MoO3納米網。在保護性氣氛中將合成的MoO3前驅體煅燒處理就得到了目標材料MoO2。采用兩相界面自組裝法合成的納米網狀材料具有比表面積大、孔隙豐富和結構穩定等優點,可以增加鋰離子的嵌入位點,提高可逆容量,同時其網狀多孔的結構可有效緩解金屬氧化物材料在充放電過程中的體積膨脹,防止活性顆粒團聚,從而利于電極循環穩定性。本原理不只適用于MoO2,也適用于其他金屬氧化物材料,形貌也不局限于本發明后文提到的幾種類型。
本發明具有如下顯著特點:
1):本發明提供了一種新的兩相界面自組裝法控制鉬酸根離子的組裝生成MoO2納米網,兩相界面種類多樣且可控,為研究者們提供新的控制材料形貌的思路。
2):本發明結合了水熱反應與磁力攪拌方式,水熱攪拌不僅提高了粒子的傳質速率和表面反應速率,同時一維的納米線在動態的反應體系中受力不均從而彎曲有利于納米網的生成。此法為三維網狀結構材料的合成提供了新的方法。
3):合成的三維網狀結構MoO2納米材料具有很高的比表面積和豐富的孔隙結構,有利于離子和電子的快速傳輸;同時該結構非常穩定,在循環過程中能有效防止活性材料的團聚和電極的粉化,進而顯著提高材料的循環性能。
本發明的積極效果:
本發明提出的兩相界面組裝法可為研究者設計和控制材料的形貌和結構提供新思路。同時,采用界面法和水熱攪拌相結合方式制備三維網狀納米結構,對研究形貌和結構對材料電化學性能的影響提供了借鑒。考慮到兩相界面和金屬氧化物種類多樣,該方法也可拓展到一些比較注重材料形貌控制的領域。
附圖說明
圖1(a),(b)分別是實施例1制備得到的MoO3前驅體的SEM圖。
圖2是實施例1制備得到的MoO3前驅體的XRD圖。
圖3(a),(b)分別是實施例1制備得到MoO2材料的SEM圖。
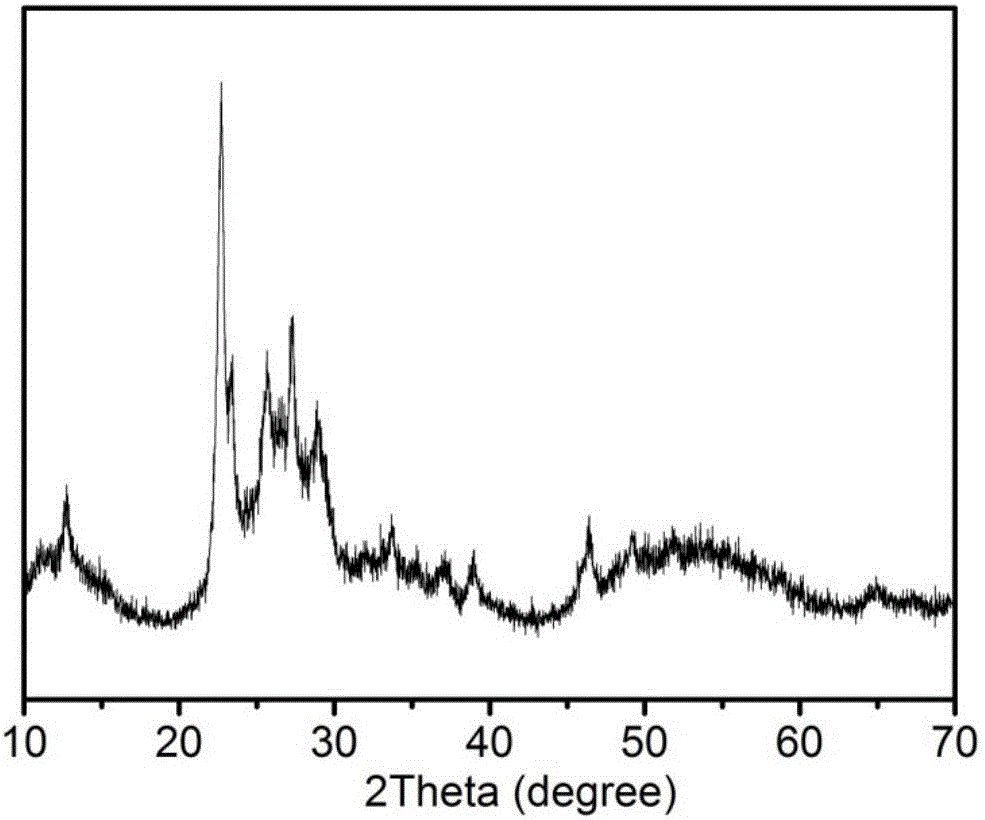
圖4是實施例1制備得到MoO2材料的XRD圖。
圖5(a),(b),(c)和(d)分別是實施例2,例3,例4和例5制備得到的MoO3前驅體的SEM圖。
具體實施方式
以下通過實施例對本發明作進一步說明,而非限制本發明。
實施例1
稱取0.747g鉬酸鈉和0.4375g檸檬酸,溶于20ml水中,用濃鹽酸調節溶液pH值為2,然后再加入20ml甲苯,攪拌均勻,將混合溶液加入帶磁力攪拌裝置的高溫反應釜中,升溫至190℃反應12h,水熱反應過程控制攪拌速率為300rpm。反應結束后,將反應釜自然冷卻至室溫,將產物離心分離,并用無水乙醇和去離子水洗滌2次后,置于烘箱中70℃干燥過夜,研磨得到MoO3前驅體。將上述深藍色前驅體置于充滿Ar氣的管式爐中,420℃下煅燒30min,然后自然冷卻至室溫,得到MoO2。圖1a和圖1b記錄了前驅體MoO3的SEM圖,可以看到MoO3為三維的納米網狀形貌,其納米網由直徑達40-70nm寬,長度達數微米的一維納米線相互交織而成。圖2記錄了前驅體MoO3的XRD圖,由圖可知該材料物相主要為MoO3,結晶性較差。圖3a和圖3b記錄了煅燒后產物MoO2的SEM圖,可以看到其完好保存了前驅體的三維網狀納米結構。圖4記錄了目標產物的XRD,由圖可知該材料物相為MoO2,結晶性良好。表1記錄了其BET比表面積測試結果,可以看到該材料的比表面積為達326m2g-1,孔徑尺寸為10-20nm。
將制備的MoO2與乙炔黑和聚偏氟乙烯(PVDF)按質量比8:1:1稱取,混合均勻后滴入數滴N-甲基吡咯烷酮(NMP)研磨至漿狀,將其涂在銅箔上,置于真空干燥箱100℃中干燥12h后用沖片機切成圓片狀電極片。以得到的極片為正極,鋰片為對電極,l mol/L的LiPF6的混合溶劑(EC:DEC=1:1體積比)為電解液,聚丙烯微孔膜為隔膜,在充滿Ar氣氣氛的手套箱中組裝成CR2016型扣式電池。表2記錄該電池在300mA g-1電流密度下的第二圈可逆容量為762mAh g-1,160次循環后容量為675mAh g-1,在2A g-1大電流密度充放電下平均放電比容量為577mAh g-1。
實施例2
稱取0.747g鉬酸鈉和0.4375g檸檬酸,溶于20ml水中,用濃鹽酸調節pH值為0,然后再加入20ml甲苯,攪拌均勻,將混合溶液加入帶磁力攪拌裝置的高溫反應釜中,升溫至180℃反應15h,水熱反應過程控制攪拌速率為300rpm。反應結束后,將反應釜自然冷卻至室溫,將產物離心分離,并用無水乙醇和去離子水洗滌2次后,置于烘箱中70℃干燥過夜,研磨得到MoO3前驅體。圖5a記錄了前驅體MoO3的SEM圖,可以看到MoO3仍然呈三維的納米網狀形貌,但其納米網由直徑達數百納米寬長度達數微米的一維納米線相互交織而成。將上述藍色前驅體置于充滿Ar氣的管式爐中,420℃下煅燒30min,然后自然冷卻至室溫,得到MoO2。表1記錄了其BET比表面積測試結果,可以看到該材料的比表面積為達223m2g-1,孔徑尺寸為10-20nm。電池組裝及電化學性能測試見實施例1。表2記錄該電池在300mA g-1電流密度下的第二圈可逆容量為832mAh g-1,160次循環后容量為613mAh g-1,在2A g-1大電流密度充放電下平均放電比容量為513mAh g-1。
實施例3
稱取0.5g鉬酸和0.4375g檸檬酸,溶于20ml水中,用濃鹽酸調節pH值為6,然后再加入20ml二甲苯,攪拌均勻,將混合溶液加入帶磁力攪拌裝置的高溫反應釜中,升溫至190℃反應12h,水熱反應過程控制攪拌速率為300rpm。反應結束后,將反應釜自然冷卻至室溫,將產物離心分離,并用無水乙醇和去離子水洗滌2次后,置于烘箱中70℃干燥過夜,研磨得到MoO3前驅體。圖5b記錄了前驅體MoO3的SEM圖,可以看到MoO3仍然呈三維的納米網狀形貌,其一維納米線直徑約為100nm,長度為數微米。將上述藍色前驅體置于充滿Ar氣的管式爐中,600℃下煅燒2h,然后自然冷卻至室溫,得到MoO2。表1記錄了其BET比表面積測試結果,可以看到該材料的比表面積為達311m2g-1,孔徑尺寸為10-20nm。電池組裝及電化學性能測試見實施例1。表2記錄該電池在300mA g-1電流密度下的第二圈可逆容量為782mAh g-1,160次循環后容量為641mAh g-1,在2A g-1大電流密度充放電下平均放電比容量為524mAh g-1。
實施例4
稱取0.632g鉬酸銨和0.563g檸檬酸鈉,溶于20ml水中,用濃鹽酸調節pH值為1,然后再加入20ml甲苯,攪拌均勻,將混合溶液加入帶磁力攪拌裝置的高溫反應釜中,升溫至200℃反應10h,水熱反應過程控制攪拌速率為500rpm。反應結束后,將反應釜自然冷卻至室溫,將產物離心分離,并用無水乙醇和去離子水洗滌2次后,置于烘箱中70℃干燥,研磨得到MoO3前驅體。圖5c記錄了前驅體MoO3的SEM圖,可以看到MoO3仍然呈三維的納米網狀形貌,其一維納米線直徑約為50nm,長度為數微米,納米線高度交連組成了納米網。將上述藍色前驅體置于充滿Ar氣的管式爐中,420℃下煅燒30min,然后自然冷卻至室溫,得到MoO2。表1記錄了其BET比表面積測試結果,可以看到該材料的比表面積為達293m2g-1,孔徑尺寸為10-20nm。電池組裝及電化學性能測試見實施例1。表2記錄該電池在300mA g-1電流密度下的第二圈可逆容量為832mAh g-1,160次循環后容量為661mAh g-1,在2A g-1大電流密度充放電下平均放電比容量為547mAh g-1。
實施例5
稱取0.747g鉬酸鈉和0.4375g檸檬酸,溶于20ml水中,用濃鹽酸調節pH值為6,然后再加入20ml甲苯,攪拌均勻,將混合溶液加入帶磁力攪拌裝置的高溫反應釜中,升溫至180℃反應15h,水熱反應過程控制攪拌速率為300rpm。反應結束后,將反應釜自然冷卻至室溫,將產物離心分離,并用無水乙醇和去離子水洗滌2次后,置于烘箱中70℃干燥過夜,研磨得到MoO3前驅體。圖5d記錄了前驅體MoO3的SEM圖,可以看到MoO3仍然呈三維的納米網狀形貌,但同時也出現了納米顆粒。將上述前驅體置于充滿Ar氣的管式爐中,420℃下煅燒30min,然后自然冷卻至室溫,得到MoO2。表1記錄了其BET比表面積測試結果,可以看到該材料的比表面積為達364m2g-1,孔徑尺寸為10-20nm。電池組裝及電化學性能測試見實施例1。表2記錄該電池在300mA g-1電流密度下的第二圈可逆容量為878mAh g-1,160次循環后容量為603mAh g-1,在2Ag-1大電流密度充放電下平均放電比容量為521mAh g-1。
實施例6
稱取0.747g鉬酸鈉和0.4375g檸檬酸,溶于20ml水中,用濃鹽酸調節pH值為2,然后再加入20ml甲苯,攪拌均勻,將混合溶液加入帶磁力攪拌裝置的高溫反應釜中,升溫至180℃反應15h,水熱反應過程控制攪拌速率為300rpm。反應結束后,將反應釜自然冷卻至室溫,將產物離心分離,并用無水乙醇和去離子水洗滌2次后,置于烘箱中70℃干燥過夜,研磨得到MoO3前驅體。將上述藍色前驅體置于充滿Ar氣的管式爐中,600℃下煅燒2h,然后自然冷卻至室溫,得到MoO2。表1記錄了其BET比表面積測試結果,可以看到該材料的比表面積為達245m2g-1,孔徑尺寸為12-20nm。電池組裝及電化學性能測試見實施例1。表2記錄該電池在300mAg-1電流密度下的第二圈可逆容量為709mAh g-1,160次循環后容量為551mAh g-1,在2Ag-1大電流密度充放電下平均放電比容量為532mAh g-1。
實施例7
稱取0.747g鉬酸鈉和0.4375g檸檬酸,溶于20ml水中,用濃鹽酸調節pH值為2,然后再加入20ml甲苯,攪拌均勻,將混合溶液加入帶磁力攪拌裝置的高溫反應釜中,升溫至180℃反應24h,水熱反應過程控制攪拌速率為100rpm。反應結束后,將反應釜自然冷卻至室溫,將產物離心分離,并用無水乙醇和去離子水洗滌2次后,置于烘箱中70℃干燥過夜,研磨得到MoO3前驅體。將上述前驅體置于充滿Ar氣的管式爐中,420℃下煅燒30min,然后自然冷卻至室溫,得到MoO2。表1記錄了其BET比表面積測試結果,可以看到該材料的比表面積為達291m2g-1,孔徑尺寸為12-25nm。電池組裝及電化學性能測試見實施例1。表2記錄該電池在300mA g-1電流密度下的第二圈可逆容量為781mAh g-1,160次循環后容量為531mAh g-1,在2Ag-1大電流密度充放電下平均放電比容量為514mAh g-1。
實施例8
稱取0.747g鉬酸鈉和0.6g抗壞血酸,溶于20ml水中,用濃鹽酸調節pH值為1,然后再加入20ml甲苯,攪拌均勻,將混合溶液加入帶磁力攪拌裝置的高溫反應釜中,升溫至180℃反應15h,水熱反應過程控制攪拌速率為400rpm。反應結束后,將反應釜自然冷卻至室溫,將產物離心分離,并用無水乙醇和去離子水洗滌2次后,置于烘箱中70℃干燥過夜,研磨得到MoO3前驅體。將上述前驅體置于充滿Ar氣的管式爐中,420℃下煅燒30min,然后自然冷卻至室溫,得到MoO2。表1記錄了其BET比表面積測試結果,可以看到該材料的比表面積為達285m2g-1,孔徑尺寸為10-25nm。電池組裝及電化學性能測試見實施例1。表2記錄該電池在300mA g-1電流密度下的第二圈可逆容量為732mAh g-1,160次循環后容量為562mAh g-1,在2Ag-1大電流密度充放電下平均放電比容量為533mAh g-1。
表1分別記錄了實施例1,例2,例3,例4,例5,例6,例7,例8制備得到的MoO2材料的BET比表面積和孔徑分布數據。
表2分別記錄了實施例1,例2,例3,例4,例5,例6,例7,例8制備得到的MoO2材料在300mA g-1循環第二圈放電容量,160次循環后的放電容量和2A g-1下平均放電容量。
表1
表2
- 還沒有人留言評論。精彩留言會獲得點贊!