一種用于陶瓷基板的高純度氮化鋁粉末材料及其制備方法與流程
本發明涉及氮化鋁材料及其制備方法,具體地說是一種用于陶瓷基板的高純度氮化鋁粉末材料及其制備方法。
背景技術:
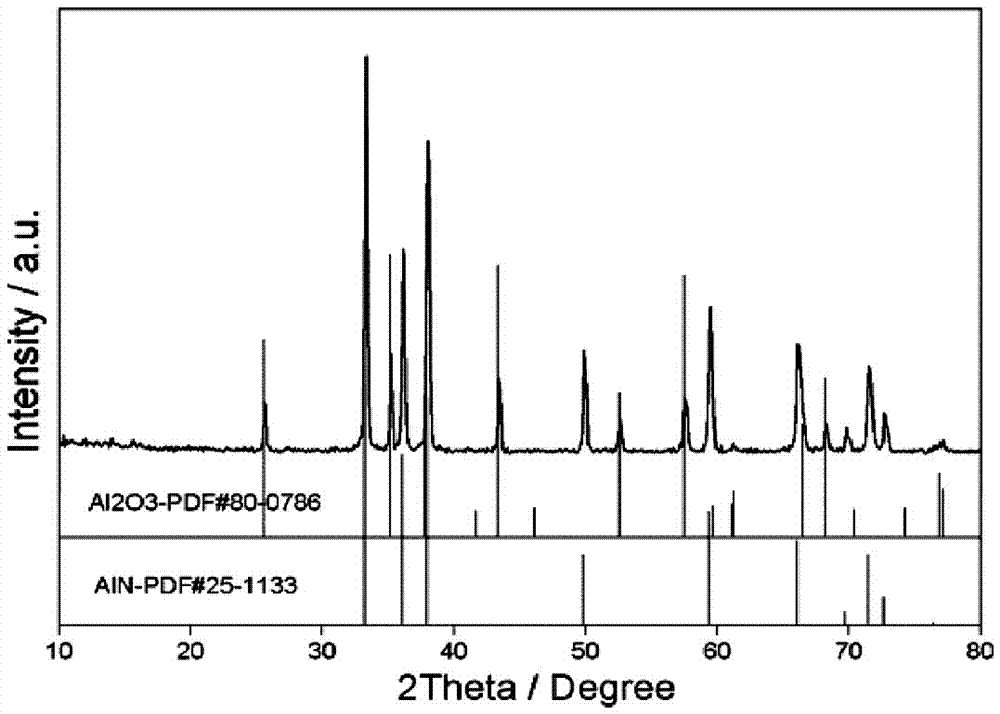
:氮化鋁陶瓷以其具有高的熱導率、與硅媲美的熱膨脹系數(2.65×10-6·k-1)、同時兼具高強度、高耐腐蝕性、高電絕緣性、低電容率和低介電損耗等優良特性,成為現代電子工業中理想的封裝和陶瓷基板材料。目前,氮化鋁粉末的制備工藝主要有以下幾種:1、鋁粉直接氮化法。高溫氮氣中,鋁粉直接與氮氣反應生成氮化鋁粉。該方法雖然工藝簡單,但是工藝過程中存在團聚嚴重現象,制備的氮化鋁粉體純度低、顆粒均勻性差。2、自蔓延高溫合成法。自蔓延高溫合成法制備氮化鋁一般采用鋁粉在高壓氮氣中被外界熱源點燃,金屬鋁與氮氣之間高化學反應使反應自維持下去,直至鋁粉完全轉化為氮化鋁。該方法可以制備高純度的氮化鋁粉,但工藝需要較高的氮氣壓力,使得成本很高,難以達到大規模生產及推廣應用。3、化學氣相沉積法。化學氣相沉積法制備氮化鋁是基于鋁的揮發性化合物與氨氣或氮氣的化學反應。該方法可以制備純度高、粒度均勻細小的氮化鋁粉,但生產效率低,所用鋁源揮發性化合物的原料價格較高,目前尚難在工業上推廣應用。4、碳熱還原氮化法。其方法是將鋁源化合物與超細碳粉充分混合,在氮氣氛圍下經高溫合成氮化鋁粉。該工藝條件要求低,采用的原料成本低,其與前幾種方法相比更適合大規模生產和應用。但是,目前的碳熱還原氮化法制備的氮化鋁粉末還存在純度低、粒徑大、顆粒團聚嚴重等缺陷,無法滿足陶瓷基板和封裝材料要求氮化鋁的粒徑小于2μm和高純度的要求。如日本德山株式會社在cn201480006112中公布了碳熱還原法制備氮化鋁粉末的方法,其氮化鋁粉末就存在顆粒團聚嚴重、粒徑大的缺陷;又如上海理工大學在cn201110193372.7公開了借助高能球磨低溫碳熱還原法制備氮化鋁,但是該方法制備的氮化鋁同樣存在粒徑大、顆粒團聚現象嚴重的問題。因此,為滿足陶瓷基板及封裝材料的使用需求,探究更好的碳熱還原工藝方法制備出低團聚、低氧含量、高純度、小粒徑的氮化鋁是行業內積極研究的課題之一。技術實現要素:本發明的目的是提供一種用于陶瓷基板的高純度氮化鋁粉末材料及其制備方法,以解決現有工藝制備的氮化鋁粉末存在純度低、粒徑大、團聚現象嚴重的問題。本發明的目的是通過以下技術方案實現的:一種用于陶瓷基板的高純度氮化鋁粉末材料,所述氮化鋁中氮含量為33.0-34.1wt%,含氧量為小于1wt%,平均粒徑為1.0-2.0μm;所述氮化鋁粉末材料是通過以下方法制備:(a)按氫氧化鋁與硼酸、燒結助劑按質量比為96-98:1-2:1-2稱量,混合均勻后轉移到馬弗爐,在600℃下退火60min,冷卻至室溫,機械研磨,得多孔al2o3粉末;所述燒結助劑為caf2、y2o3或nh4f中的任意一種;(b)多孔al2o3粉末與去離子水按質量體積比為66.7g:200ml混合,攪拌,水浴加熱至60℃時,加入無機鋁鹽和表面活性劑,混合均勻,用稀鹽酸調節溶液的ph值至2-4,得懸浮液;所述多孔al2o3粉末、無機鋁鹽和表面活性劑的質量體積比為66.7g:5g:0.5ml;所述無機鋁鹽為al(no3)3和alcl3中的至少一種;所述表面活性劑為聚乙二醇;(c)在所述懸浮液中加入超細碳粉,攪拌均勻,逐滴加入氨水至懸浮液的ph值到10-13停止滴加,60℃水浴,攪拌,直至懸浮液變為均勻黑色膠狀物;所述超細碳粉的添加量為所述多孔al2o3粉末質量的50%;(d)將所述黑色膠狀物真空干燥,將干燥后的黑色塊狀物研磨至粉末狀,將黑色粉末移到石墨坩堝中并置于碳管爐中,抽真空后通入氮氫混合氣流,在1550-1700℃反應5-7h,得黑色固體;(e)將所述黑色固體研磨,置于溫度為650-750℃下保溫脫碳1-3h即得。本發明提供的用于陶瓷基板的高純度氮化鋁粉末材料制備方法,包括以下步驟:(a)按氫氧化鋁與硼酸、燒結助劑按質量比為96-98:1-2:1-2稱量,混合均勻后轉移到馬弗爐,在600℃下退火60min,冷卻至室溫,機械研磨,得多孔al2o3粉末;所述燒結助劑為caf2、y2o3或nh4f中的任意一種;(b)多孔al2o3粉末與去離子水按質量體積比為66.7g:200ml混合,攪拌,水浴加熱至60℃時,加入無機鋁鹽和表面活性劑,混合均勻,用稀鹽酸調節溶液的ph值至2-4,得懸浮液;所述多孔al2o3粉末、無機鋁鹽和表面活性劑的質量體積比為66.7g:5g:0.5ml;所述無機鋁鹽為al(no3)3和alcl3中的至少一種;所述表面活性劑為聚乙二醇;(c)在所述懸浮液中加入超細碳粉,攪拌均勻,逐滴加入氨水至懸浮液的ph值到10-13停止滴加,60℃水浴,攪拌,直至懸浮液變為均勻黑色膠狀物;所述超細碳粉的添加量為所述多孔al2o3粉末質量的50%;(d)將所述黑色膠狀物真空干燥,將干燥后的黑色塊狀物研磨至粉末狀,將黑色粉末移到石墨坩堝中并置于碳管爐中,抽真空后通入氮氫混合氣流,在1550-1700℃反應5-7h,得黑色固體;(e)將所述黑色固體研磨,置于溫度為650-750℃下保溫脫碳1-3h即得高純度氮化鋁粉末。本發明制備方法中步驟(c)所述超細碳粉的粒徑為10-40nm。本發明制備方法中步驟(d)的氮氫混合氣流中氮氣和氫氣的體積比為9:1,所述氮氫混合氣流的流速為5-10l/h。本發明通過特定的工藝步驟首次制備了純度高、粒徑小、顆粒分布窄、氧含量低的氮化鋁粉末,本發明制備的氮化鋁粒徑可以控制在0.1-0.2μm,純度高,含氧量可控制在1wt%,能夠滿足陶瓷基板燒結的要求,可與其他添加劑形成均勻穩定的漿料,進而制備致密度高、導熱率高氮化鋁陶瓷基板;純度高、粒徑小可提高燒結基板的致密度;氧含量低可有效避免氮化鋁晶格中鋁空位的數量,進而提高燒結基板的熱導率;而且本發明提供制備過程簡單,操作成本低,工藝窗口寬,適于大規模工業化生產,所制備的高純氮化鋁粉末可作為陶瓷基板及封裝材料廣泛使用。附圖說明圖1是實施例30制備的氮化鋁粉末材料的xrd圖。圖2是對比例1制備的氮化鋁粉末材料的xrd圖。圖3是實施例30制備的氮化鋁粉末材料的sem圖。圖4是對比例1制備的氮化鋁粉末材料的sem圖。具體實施方式下面實施例用于進一步詳細說明本發明,但實施例并不對本發明做任何形式的限定。除非特別說明,本發明采用的試劑、方法和設備為本
技術領域:
常規試劑、方法和設備。但不以任何形式限制本發明。實施例1(1)分別稱取98g氫氧化鋁、1g硼酸和1gcaf2,混合放入瑪瑙研缽中,研磨30min,將混合均勻的粉末樣品導入剛玉坩堝中,將剛玉坩堝轉移到馬弗爐,600℃退火1h;待馬弗爐自然冷卻至室溫,將燒結得到的al2o3樣品取出,機械研磨10min,得多孔al2o3粉末;(2)將66.7g的多孔al2o3粉末倒入裝有200ml去離子水的燒杯中,將燒杯放入恒溫水浴鍋,玻璃棒攪拌,加熱升溫到60℃;向溶液中加入5g硝酸鋁,繼續攪拌,待硝酸鋁完全溶解后,加入0.5ml聚乙二醇,用稀鹽酸調節溶液至ph值=2,得懸浮液;(3)向燒杯中的懸浮液中加入33.3g平均粒徑為10nm的碳粉,60℃水浴攪拌,待攪拌均勻后,緩慢滴加氨水至溶液的ph值=12,停止滴加,繼續水浴(60℃)攪拌,直至燒杯里物質變成均勻的黑色膠狀物;(4)將盛有黑色膠狀物的燒杯放入真空干燥箱,在80℃條件下真空保溫干燥12h,將得到的黑色塊狀固體轉移到瑪瑙研缽中研磨,將研磨得到的黑色固體粉末轉移到石墨坩堝,將石墨坩堝放入碳管爐,抽真空3次后,通入氮氫混合氣流,氮氣和氫氣的體積比為9:1,其氮氫混合氣流的流速為10l/h,在1550℃條件下,保溫5h,冷卻至室溫后,得黑色固體;(5)將黑色固體研磨為黑色粉末,從石墨坩堝轉移到剛玉坩堝,將剛玉坩堝放入箱式爐,在空氣流量10l/min,700℃條件下保溫1h,即得高純度氮化鋁粉末材料。實施例2-12實施例2-12的制備方法同實施例1,僅改變了步驟(1)中的硼酸的添加量和助劑的種類。實施例13(1)分別稱取97g氫氧化鋁、2g硼酸和1gy2o3,混合放入瑪瑙研缽中,研磨30min,將混合均勻的粉末樣品導入剛玉坩堝中,將剛玉坩堝轉移到馬弗爐,600℃退火1h;待馬弗爐自然冷卻至室溫,將燒結得到的al2o3樣品取出,機械研磨10min,得多孔al2o3粉末;(2)將66.7g多孔al2o3粉末倒入裝有200ml去離子水的燒杯中,將燒杯放入恒溫水浴鍋,玻璃棒攪拌,加熱升溫到60℃;向溶液中加入5g硝酸鋁,繼續攪拌,待硝酸鋁完全溶解后,加入0.5ml聚乙二醇,用稀鹽酸調節溶液至ph值=2,得懸浮液;(3)向燒杯中的懸浮液中加入33.3g平均粒徑為10nm的碳粉,繼續水浴(60℃)攪拌,待攪拌均勻后,緩慢滴加氨水至溶液的ph值=12,停止滴加,繼續水浴(60℃)攪拌,直至燒杯里物質變成均勻的黑色膠狀物;(4)將盛有黑色膠狀物的燒杯放入真空干燥箱,在80℃條件下真空保溫干燥12h,將得到的黑色塊狀固體轉移到瑪瑙研缽中研磨,將研磨得到的黑色固體粉末轉移到石墨坩堝,將石墨坩堝放入碳管爐,抽真空3次后,通入氮氫混合氣流,氮氣和氫氣的體積比為9:1,其氮氫混合氣流的流速為10l/h,在1550℃條件下,保溫5h,冷卻至室溫后,得黑色固體;(5)將黑色固體研磨為黑色粉末,從石墨坩堝轉移到剛玉坩堝,將剛玉坩堝放入箱式爐,在空氣流量10l/min,700℃條件下保溫1h,即得高純度氮化鋁粉末材料。實施例14-18實施例14-18的制備方法與實施例13一致,僅改變了步驟(2)中的無機鋁鹽的種類和添加比例。編號al(oh)3/g硼酸/gy2o3/g硝酸鋁/g氯化鋁/g實施例14972141實施例15972132實施例16972123實施例17972114實施例18972105實施例19-20實施例19-20的制備方法與實施例16一致,僅改變了步驟(2)中用稀鹽酸調節溶液后的ph值。實施例21-23實施例21-23的制備方法與實施例16一致,僅改變了步驟(3)中用碳粉的粒徑。實施例24-26實施例24-26的制備方法與實施例16一致,僅改變了步驟(3)中用氨水調節溶液的ph值。實施例27-32實施例27-32的制備方法與實施例25一致,改變了步驟(4)中的氮化溫度以及氮氫混合氣體的通入量。實施例33-36實施例33-36的制備方法與實施例30一致,改變了步驟(5)中的脫碳溫度和空氣流量。對比例1根據專利cn201380009981.x中公開的技術內容,按照其公開的實施例1進行重復性實驗。將平均粒徑1.2μm、比表面積10.7m2/g的α-al2o3及比表面積125m2/g的炭黑與平均粒徑1μm的氧化釔混合;將上述均勻混合的粉末在1700℃和80%n2/co6l/min條件下,保溫10h;將氮化后的粉末樣品空氣氣氛下700℃氧化處理5h除去過量的炭黑,制得氮化鋁粉末。實施例37對實施例和對比例制備的氮化鋁粉末進行性能檢測,檢測指標有:氮含量測試;氧含量測試;平均粒徑檢測以及k值((d90-d10)/d50);氮含量以及氧含量結果由氧氮分析儀測試得出,平均粒徑由粒度儀進行檢測。以實施例30為例,對實施例30和對比例1制備的氮化鋁粉末材料檢測得xrd圖如圖1和圖2所示;對實施例30和對比例1制備的氮化鋁粉末材料測得的sem圖如圖3和圖4所示。檢測結果見表1。表1實施例1-36以及對比例1-14的性能由表1可知,實施例34選擇y2o3作為助劑,al(oh)3:硼酸:y2o3=97:2:1,硝酸鋁:氯化鋁=2:3,溶液(稀hcl調節)ph值為2,炭黑粒徑為10nm,溶液(氨水調節)ph值為11,氮化溫度為1600℃,氮氣流速為10l/min,脫碳溫度650℃,空氣流量15l/min,氮化率為100%,制得的氮化鋁平均粒徑最小,為1.0μm,含氧量最小,為0.7%。上述實施例為本發明較佳的實施方式,但本發明的實施方式并不受所述實施例的限制,其他的任何未背離本發明的精神實質與原理下所作的改變、修飾、替代、組合、簡化,均應為等效的置換方式,都包含在本發明的保護范圍之內。當前第1頁12
相關技術
網友詢問留言
已有0條留言
- 還沒有人留言評論。精彩留言會獲得點贊!
1