4位含有取代基的2?(2`?羥基苯基)苯并唑化合物及其制法和用途的制作方法
本發明屬于有機半導體光電技術領域,具體涉及一種4位含有取代基的2-(2'-羥基苯基)苯并唑化合物及其制備和應用。
背景技術:
1987年,美國的C.W.Tang等人發明以真空蒸鍍法制成多層式結構的有機發光二極管(OLED),使得有機電致發光器件的研究受到各國科學家的高度重視。OLED技術的發展對光電顯示領域產生了翻天覆地的變化。如今材料科學的發展和理論工藝的成熟對新型OLED的發展起著至關重要的作用,使得材料領域的研究人員對發光材料的探索產生了極大地興趣。由于白光有機發光二極管(WOLED)具有效率高、亮度高、功耗低、視角廣、響應速度快、主動發光、超薄超輕以及可柔性化等優異性能,并在顯示和照明領域有廣闊應用前景,受到學者和業界的廣泛重視而成為研究熱點。
1994年,日本山形大學的Kido教授等首先制備了世界上第一個WOLED器件,盡管當時器件的最大功率效率只有0.83lm/W,最大亮度只有3400cd/M2,但是還是在有機電致發光器件領域實現了突破性的進展。
2003年,WOLED器件效率達到了15lm/W并首次超過了白熾燈。2006年,美國圣地亞哥大學的Nobuhiro Ide研究小組利用全磷光材料制備出顏色可調控的堆疊式多層發光結構的WOLED,其器件效率達到了62.8lm/W。2008年,美國圣地亞哥大學的James Esler研究小組制備出最大功率效率為100 lm/W多發光層結構的WOLED,其工作電壓為3.6V,外量子效率達到了20%。
多發光層WOLED的效率通常較高,但是結構和工藝條件相對復雜、電壓較高,導致成本也相對較高,各發光層之間存在異質結,并且色坐標穩定性一般較差。如何合理地控制激子產生區域,激子復合區域,各發光層之間能量轉移和激子擴散是獲得高性能的保證。
目前,在WOLED領域,開發一種器件效率高、工藝簡單,成本低,穩定性好的單層白光器件是實現白光有機發光二極管商業化的關鍵。
技術實現要素:
針對目前白光有機發光二極管中存在的器件制備工藝復雜、制作成本較高、重復性較差等技術問題,本發明提供了一種新的有機熒光材料,即4位含有取代基的2-(2'-羥基苯基)苯并唑化合物,及其制法和應用。所述的有機熒光材料是一系列具有激發態分子內質子轉移化合物的四能級躍遷性質的2-(2'-羥基苯基)苯并唑,并連接多種含芳烴共軛結構單元的新材料。這類材料具有熒光量子產率高,斯托克位移大,合成簡單,產率高等優點。通過混合商業化的藍光材料組成二元互補色,可以制備工藝簡單的單層白光器件。本發明采用的技術方案如下。
本發明提供了一種4位含有取代基的2-(2'-羥基苯基)苯并唑化合物。
本發明所述的4位含有取代基的2-(2'-羥基苯基)苯并唑化合物,其結構通式為:
式中:X為S、O或NH,Ar為含芳烴共軛結構單元。
所述含芳烴共軛結構單元選自以下取代基結構中的一種:
本發明還提供上述4位含有取代基的2-(2'-羥基苯基)苯并唑化合物的制備方法。
本發明所述的4位含有取代基的2-(2'-羥基苯基)苯并唑化合物的制備方法,其包括如下步驟:
(1)含芳烴共軛結構化合物的硼酸化:在氮氣保護下,溴代芳烴溶于干燥的有機溶劑,溫度低于-40℃條件下加入正丁基鋰,攪拌2到4小時后,加入硼酸化合物,硼酸化,升溫到常溫后反應15-20小時。反應結束后,萃取有機相,將有機相合并干燥,得到產物。
(2)將步驟(1)的產物和2-(2'-4-溴苯酚)苯并唑,鈀催化劑溶于有機溶劑,然后加入強堿水溶液,氮氣保護下反應。反應結束后,萃取有機相,合并有機相并濃縮,分離提純后,得到終產物,即為本發明的產物。
上述步驟(1)中,低溫反應可使用干冰丙酮浴冷卻;所述硼酸化合物為硼酸正三丁酯;使用水和二氯甲烷萃取有機相;所述有機溶劑為四氫呋喃。
上述步驟(2)中,所述鈀催化劑為四(三苯基膦)鈀、雙(二亞芐基丙酮)鈀、1,1-雙(二苯膦基)二茂鐵二氯,強堿為碳酸鉀、氯化鉀、氟化鉀中的一種或幾種,所述有機溶劑為四氫呋喃和甲苯的體積比為1:1的混合溶劑。反應溫度為80-120℃,反應時間為24-60小時。所述的2-(2'-4-溴苯酚)苯并唑化合物的通式為:
其中,X為S、O或NH。
上述制備方法中,按摩爾比計,步驟(1)中,溴代芳烴和正丁基鋰的比例為1:0.9~1.5,優選1:1.2;步驟(2)中,所述硼酸化溴代芳烴和2-(2'-4-溴苯酚)苯并唑的比例為1:0.5~1.5,優選1:1
所述溴代芳烴和有機溶劑的添加比例為1摩爾:2~8升。優選比例為1摩爾:4~6升。
本發明的再一目的在于提供基于4位含有取代基的2-(2'-羥基苯基)苯并唑化合物用于制備白光有機發光二極管的用途。
本發明所述的4位含有取代基的2-(2'-羥基苯基)苯并唑化合物可以作為發光材料,用于有機白光二極管器件中。通過混合商業化的藍光材料組成二元互補色,可以制備工藝簡單的單層白光器件。
本發明具有如下有益效果:1、本發明的4位含有取代基的2-(2'-羥基苯基)苯并唑化合物具有熒光量子產率高,斯托克位移大;2、上述化合物的制備方法合成簡單,產率高,利于擴大生產規模;3、上述化合物可以作為發光材料,用于有機白光二極管器件中。
附圖說明
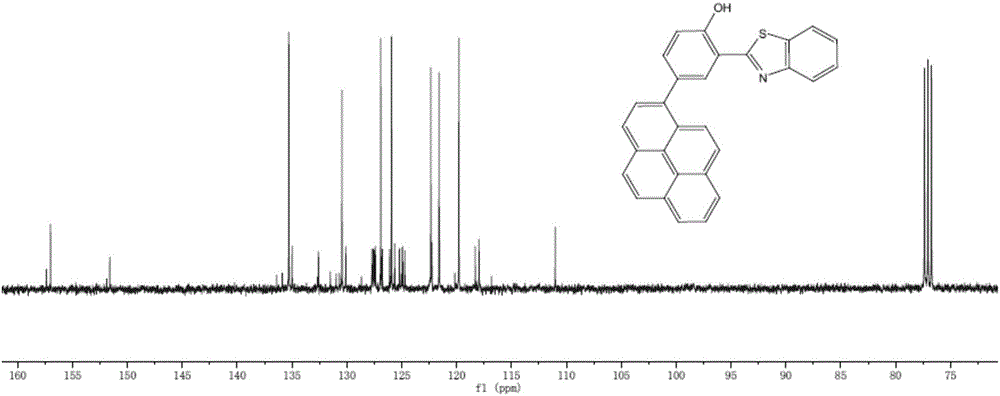
圖1本發明實施例2化合物的碳譜圖;
圖2本發明實施例3化合物的碳譜圖;
圖3本發明實施例2化合物的熒光吸收-發射圖;
圖4本發明實施例3化合物的熒光吸收-發射圖;
圖5本發明實施例2化合物的氧化還原曲線。
具體實施方式
為了更好理解本發明專利的內容,下面通過具體的實例來進一步說明本發明的技術方案。但這些實施實例并不限制本發明。
該材料2-(2'-4-溴苯酚)苯并唑通過鈴木反應連接多種芳烴共軛結構單元的給電子基團,該材料具有如下的通式:
式中:X為S、O或NH。
實施例1、
首先,2-(2'-4-溴苯酚)苯并噻唑的制備:
取二氨基酚(4.4g,20mmol,1equiv)和對氨基苯甲酸(2.5g,20mmol,1equiv)加入130ml多聚磷酸中,在N2的環境中加熱到168℃,并回流6小時。待反應完全后,冷卻后將產物溶于100ml冰水混合物中,向其中加入飽和的NaOH水溶液中和。待pH值調為6左右改加NaHCO3,邊加邊攪拌,到不再有CO2氣泡產生。將中和后的溶液靜置待溶液澄清后抽濾。把抽濾出來的固體加入飽和NaHCO3水溶液用乙酸乙酯萃取三次,合并有機層,用無水Na2SO4干燥旋蒸后得到綠色產物5g,產率為83%。(下述實施例中,2-(2'-4-溴苯酚)苯并噻唑的制備方法都與此相同,不再贅述)。
其次,2-(2'-苯并噻唑)-4-(9'-菲)苯酚的制備
(1)含芳烴共軛結構化合物的硼酸化:在氮氣保護下,9-溴代菲溶于干燥的四氫呋喃,置于干冰丙酮浴(-78℃)中冷卻,低溫下加入正丁基鋰,9-溴代菲和正丁基鋰的添加比例為1:0.9攪拌2小時后,加入硼酸正三丁脂,硼酸化,繼續反應0.5小時,撤去干冰丙酮浴,升溫到常溫后反應15小時。反應結束后,在冰水浴下淬滅反應,然后用水和二氯甲烷萃取有機相,將有機相合并干燥旋干,得到產物。
(2)取9-硼酸菲(0.18g,0.75mmol,1當量)和2-(2'-4-溴苯酚)苯并噻唑(0.12g,0.38mmol,0.5當量)混合溶于20ml四氫呋喃和甲苯的體積比為1:1的混合溶液中。加入催化劑Pd(pph3)4。避光通氮氣,再加入10ml濃度為2mol/L的碳酸鉀和氟化鉀的水溶液。在80℃的條件下反應24小時。反應結束后,萃取有機相,合并有機相并濃縮,通過層析柱的方法進行分離提純,得到黃色粉末狀終產物。
實施例2、
2-(2'-苯并噻唑)-4-(9'-菲)苯酚的制備
(1)含芳烴共軛結構化合物的硼酸化:在氮氣保護下,9-溴代菲溶于干燥的四氫呋喃,置于干冰丙酮浴中冷卻至-78℃,低溫下加入正丁基鋰,9-溴代菲和正丁基鋰的添加比例為1:1.2,攪拌3小時后,加入硼酸正三丁脂,硼酸化,繼續反應0.5小時,撤去干冰丙酮浴,升溫到常溫后反應18小時。反應結束后,在冰水浴下淬滅反應,然后用水和二氯甲烷萃取有機相,將有機相合并干燥旋干,得到產物。
(2)取9-硼酸菲(0.18g,0.75mmol,1當量)和2-(2'-4-溴苯酚)苯并噻唑(0.24g,0.75mmol,1當量)混合溶于20ml四氫呋喃和甲苯的體積比為1:1的混合溶液中。加入催化劑Pd(pph3)4。避光通氮氣,再加入10ml濃度為2mol/L的碳酸鉀和氟化鉀的水溶液。在90℃的條件下反應48小時。反應結束后,萃取有機相,合并有機相并濃縮,通過層析柱的方法進行分離提純,得到黃色粉末狀終產物0.31g,產率為82%。
對終產物進行核磁共振和氧化還原曲線檢測,分別如圖1和圖5所示
實施例3、
2-(2'-苯并噻唑)-4-(1'-芘)苯酚的制備
將實施例2中的9-硼酸菲換成1-硼酸芘,具體步驟為:
(1)含芳烴共軛結構化合物的硼酸化:在氮氣保護下,1-溴代芘溶于干燥的四氫呋喃,置于干冰丙酮浴中冷卻至-78℃,低溫下加入正丁基鋰,1-溴代芘和正丁基鋰的添加比例為1:1.2,攪拌3小時后,加入硼酸正三丁脂,硼酸化,繼續反應0.5小時,撤去干冰丙酮浴,升溫到常溫后反應18小時。反應結束后,在冰水浴下淬滅反應,然后用水和二氯甲烷萃取有機相,將有機相合并干燥旋干,得到產物。
(2)取9-硼酸芘(0.6g,2.2mol,1當量)和2-(2'-4-溴苯酚)苯并噻唑(0.7g,2.2mmol,1當量)混合溶于30ml四氫呋喃和甲苯的體積比為1:1的混合溶液中。加入催化劑Pd(pph3)4。避光通氮氣,再加入10ml濃度為2mol/L的碳酸鉀和氟化鉀的水溶液。在90℃的條件下反應48小時。反應結束后,萃取有機相,合并有機相并濃縮,通過層析柱的方法進行分離提純,得到黃色粉末狀終產物0.86g,產率為73%。
對終產物進行核磁共振檢測,如圖2所示。
實施例4、
2-(2'-苯并噻唑)-4-(1'-芘)苯酚的制備
(1)含芳烴共軛結構化合物的硼酸化:在氮氣保護下,1-溴代芘溶于干燥的四氫呋喃,置于干冰丙酮浴中冷卻至-78℃,低溫下加入正丁基鋰,1-溴代芘和正丁基鋰的添加比例為1:1.5,攪拌4小時后,加入硼酸正三丁脂,硼酸化,繼續反應0.5小時,撤去干冰丙酮浴,升溫到常溫后反應20小時。反應結束后,在冰水浴下淬滅反應,然后用水和二氯甲烷萃取有機相,將有機相合并干燥旋干,得到產物。
(2)取9-硼酸芘(0.6g,2.2mol,1當量)和2-(2'-4-溴苯酚)苯并噻唑(0.36g,3.3mmol,1.5當量)混合溶于30ml四氫呋喃和甲苯的體積比為1:1的混合溶液中。加入催化劑Pd(pph3)4。避光通氮氣,再加入10ml濃度為2mol/L的碳酸鉀和氟化鉀的水溶液。在120℃的條件下反應60小時。反應結束后,萃取有機相,合并有機相并濃縮,通過層析柱的方法進行分離提純,得到黃色粉末狀終產物。
實施例5、
3-(2'-苯并噻唑)-4'-(1,2,2-三苯乙烯)-4'-[1,1'-聯苯]的制備
(1)含芳烴共軛結構化合物的硼酸化:在氮氣保護下,溴代四苯乙烯溶于干燥的四氫呋喃,置于干冰丙酮浴中冷卻至-78℃,低溫下加入正丁基鋰,溴代四苯乙烯和正丁基鋰的添加比例為1:1.2,攪拌3小時后,加入硼酸正三丁脂,硼酸化,繼續反應0.5小時,撤去干冰丙酮浴,升溫到常溫后反應18小時。反應結束后,在冰水浴下淬滅反應,然后用水和二氯甲烷萃取有機相,將有機相合并干燥旋干,得到產物。
(2)取硼酸四苯乙烯(0.5g,1.9mol,1當量)和2-(2'-4-溴苯酚)苯并噻唑(0.65g,1.9mmol,1當量)混合溶于40ml四氫呋喃和甲苯的體積比為1:1的混合溶液中。加入催化劑Pd(pph3)4。避光通氮氣,再加入10ml濃度為2mol/L的碳酸鉀和氟化鉀的水溶液。在90℃的條件下反應48小時。反應結束后,萃取有機相,合并有機相并濃縮,通過層析柱的方法進行分離提純,得到淡黃色粉末狀終產物。
實施例6、
本實施例是對實施例2和實施例3制備的化合物光譜的測定。
將實施例2和實施例3的化合物配成標準的1μM的四氫呋喃溶液。采用島津-3150紫外可見光譜儀和RF-520XPC熒光光譜儀進行吸收光譜和發射光譜的測定。光致發光光譜是在紫外吸收的最大吸收出測得的。如圖3和圖4所示,由圖可知,兩個化合物的吸收和熒光重疊范圍很小甚至不存在重疊,說明這兩個材料有著斯托克位移大的優點。
實施例6、
本實施例是對實施例2和實施例3制備的化合物電化學的測定。
電化學循環伏安(CV)實驗在一個Eco Chemie B.V.AUTOLABpotentiostat伏安分析儀上完成的,采用三電極體系,包括鉑碳電極、Ag/Ag+為參比電極、鉑絲為對電極。氧化過程采用二氯甲烷作為溶劑,還原過程采用四氫呋喃作為溶劑,六氟磷四丁基銨(Bu4N+PF6-)作為支持電解質,濃度為0.1M。所有電化學實驗都是在常溫下氮氣氣氛中進行的,電壓掃描速度0.1V/S。使用二茂鐵(FOC)作為基準,通過測量氧化和還原過程的開始電壓可以計算材料的HOMO和LUMO能級。
從電化學測試結果圖5可以看出,材料的氧化還原峰的位置,再通過公式:
HOMO=-[EOX–EFe/Fe++4.8]
LUMO=-[ERE–EFe/Fe++4.8]
計算可以得到實施例2材料的HOMO和LUMO能級分別為-5.44eV和-3.40eV。實施例3材料的HOMO和LUMO能級分別為-5.26eV和-3.44eV。
- 還沒有人留言評論。精彩留言會獲得點贊!
- 一種二苯并噻吩的萃取方法與流程
- 取代的苯并噻吩基衍生物作為用于治療II型糖尿病的GPR40激動劑的制造方法與工藝
- 2?哌嗪?1?基?4H?1,3?苯并噻嗪?4?酮衍生物及其用于治療哺乳動物感染的用途的制造方法與工藝
- 一種甲硫基取代苯并[d]惡唑衍生物的制備方法與流程
- 包括一種或多種1,1?二取代的烯烴化合物的乳液聚合物、乳液法及聚合物組合物的制造方法與工藝
- 包括一種或多種1,1?二取代的烯烴化合物的聚合物及其聚合物組合物的制造方法與工藝
- 包括一種或多種1,1?二取代的烯烴化合物的溶液聚合物、溶液聚合法及聚合物組合物的制造方法與工藝
- 一種異原子取代的苯并噻二唑基聚合物給體材料及其制備方法和應用與流程
- 5,7位取代的1H?苯并[e][1,4]二氮雜?2(3H)?酮衍生物的制備方法與流程
- 具有兩個二苯并呋喃或二苯并噻吩取代基的咔唑的制造方法與工藝
- 一種含硫的取代基取代的二噻吩衍生物及其共軛聚合物的制備方法及應用
- N2-糖基取代的1,2,3-三唑化合物及其合成方法與應用
- 4-(3-取代苯胺基)-6-取代基喹唑啉類化合物及制備和應用
- 被具有包含炔基取代基的苯基取代的除草活性環二酮化合物或其衍生物的制作方法
- 1-N-取代芐基-6-N’-取代基-2,3,6,9-四氫-1H-[1,4]噁嗪并[3,2-g]喹啉-9-酮-8-甲酸 ...的制作方法
- 具有取代苯基硫屬原子代低級烷基和導入酯結構的苯基作為取代基的新型1,2-二氫喹啉 ...的制作方法
- 2
- 制備區域規則的聚-(3-取代的)噻吩、硒吩、噻唑和硒唑的方法
- 一種取代五并苯類紅色有機電致發光材料及其制備方法
- 光子型間苯取代基全氟環戊烯類二芳烯光致變色材料的制備及其在雙光子光存儲中的應用的制作方法