一種基于氧化吡啶配體的金屬有機框架及制備方法與應用與流程
本發明屬于催化劑制備技術領域,具體涉及一種基于氧化吡啶配體的金屬有機框架及制備方法與應用。
背景技術:
醛或酮與活性亞甲基化合物的脫水縮合反應,形成碳碳雙鍵的反應稱為Knoevenagel縮合反應。此類反應能夠直接合成大量有用的化合物,在工業、農業、藥學、生物科學等諸多領域有著廣泛應用。Knoevenagel縮合反應一般是用Lewis酸或堿為催化劑,亦可采用弱堿(胺、吡啶等)作催化劑,在均相或異相環境中進行反應,一般所需時間較長,而且產率較低。
金屬-有機框架材料(Metal-OrganicFrameworks,MOFs)是一類有機-無機雜化材料,由有機配體和無機金屬單元構建而成。一般具有多變的拓撲結構以及物理化學性質。與傳統的多孔材料相比,MOFs具有較大的比表面積和框架內孔體積,作為一類功能化材料在催化方面表現出很大的優勢。
然而現有金屬有機框架作為一種路易斯酸催化劑存在的問題有穩定性差、制備步驟繁瑣、活性低、不能回收等。
技術實現要素:
為克服現有技術的缺陷,本發明提供了一種基于氧化吡啶配體的金屬有機框架及制備方法與應用。
為實現上述目的,本發明的技術方案為:
一種氧化吡啶配體L,其中,L的化學結構式為:
上述氧化吡啶配體L的制備方法,(1)將3,5-二鹵代吡啶制備成3,5-二鹵代吡啶-N-氧化物,(2)將3,5-二鹵代吡啶-N-氧化物經過Suzuki偶聯反應獲得氧化吡啶配體L。
所述Suzuki偶聯反應為也稱作鈴木反應,是一個較新的有機偶聯反應,零價鈀配合物催化下,芳基、吡啶或烯基硼酸(硼酸酯)與氯、溴、碘代芳烴(吡啶或烯烴)發生交叉偶聯。
3,5-二鹵代吡啶的結構式為:
其中,R為鹵代基團,如Cl、Br等。
3,5-二鹵代吡啶-N-氧化物的結構式為:
其中,R為鹵代基團,如Cl、Br等。
優選的,(1)采用間氯過氧苯甲酸將3,5-二溴吡啶制備成3,5-二溴吡啶-N-氧化物,(2)將3,5-二溴吡啶-N-氧化物與4-吡啶硼酸經過Suzuki偶聯反應獲得氧化吡啶配體L。
優選的,步驟(1)采用的溶劑為四氫呋喃。
優選的,步驟(1)中的反應條件為:在常溫下攪拌48h。
進一步優選的,步驟(1)中3,5-二溴吡啶與間氯過氧苯甲酸的摩爾比為1:2.50~3.00。
更進一步優選的,步驟(1)中3,5-二溴吡啶與間氯過氧苯甲酸的摩爾比為1:2.96。
優選的,步驟(2)采用的催化劑為四(三苯基膦)鈀。
優選的,步驟(2)的反應條件為:加熱回流48小時,加熱回流溫度為75-80℃。
優選的,步驟(2)中3,5-二溴吡啶-N-氧化物與4-吡啶硼酸的摩爾比為1:2.2~2.5。
進一步優選的,步驟(2)中3,5-二溴吡啶-N-氧化物與4-吡啶硼酸的摩爾比為1:2.4。
優選的,所述Suzuki偶聯反應中采用的堿為K2CO3。
進一步優選的,步驟(2)中3,5-二溴吡啶-N-氧化物與K2CO3的摩爾比為1:12.5。
一種金屬有機框架Zn-MOF,其結構式為(ZnLBr2)n,其中,L為上述氧化吡啶配體L,n為非零的自然數。
優選的,所述金屬有機框架是單斜晶系,屬于P2(1)/n空間群。
進一步優選的,所述金屬有機框架存在一種Zn(II)中心,其處于(ZnN2Br2)的配位環境中,和Zn(II)配位的氮原子是來自于配體L的端基吡啶,Zn-N鍵的長度分別是和其中,配體L中氧化吡啶的氧未參與配位,Zn(II)中心通過配體末端的吡啶氮原子相連,沿b軸方向伸展為一維螺旋鏈;一條鏈上氧化吡啶上的氧原子和另外一條鏈上氧化吡啶上的氫原子形成氫鍵,這些氫鍵將上述一維螺旋鏈進一步連接為波浪狀的手性二維面。
更進一步優選的,兩個Zn(II)之間的最短的距離是
一種金屬有機框架Zn-MOF的制備方法,將ZnBr2溶液鋪在上述氧化吡啶配體L溶液上,靜置后得到無色單晶即為金屬有機框架Zn-MOF。
優選的,所述ZnBr2溶液中的溶劑為甲醇。
優選的,所述ZnBr2溶液中ZnBr2的濃度為0.005mol/L。
優選的,所述氧化吡啶配體L溶液的溶劑為二氯甲烷。
優選的,所述氧化吡啶配體L溶液中氧化吡啶配體L的濃度為0.005mol/L。
優選的,ZnBr2與氧化吡啶配體L的投入摩爾比為1:1。
優選的,制備溫度為室溫。
本發明中所述的室溫為15-25℃。
一種上述金屬有機框架Zn-MOF在催化Knoevenagel縮合反應中的應用。
一種催化劑,包括上述金屬有機框架Zn-MOF。
一種芐烯丙二腈的合成方法,以苯甲醛和丙二腈為原料,以上述金屬有機框架Zn-MOF為催化劑,在室溫下反應后即得芐烯丙二腈。
優選的,所述苯甲醛和丙二腈的摩爾比為1:1.2。
優選的,其步驟為,將苯甲醛與丙二腈混合攪拌一段時間后加入催化劑,繼續攪拌一段時間后,即可獲得芐烯丙二腈。
優選的,所述苯甲醛與催化劑的摩爾比為1:0.04。
一種上述金屬有機框架Zn-MOF在催化Knoevenagel縮合反應后的回收方法,Knoevenagel縮合反應后,對混合液進行離心分離即可對金屬有機框架Zn-MOF進行回收。
本發明的有益效果:
(1)采用本發明的Zn-MOF進行催化,實現了異相催化,同時制備的Zn-MOF穩定性好,可以重復利用五次以上,并且催化劑回收容易,提高了催化劑的利用率,降低了成本。
(2)本發明的反應溫度溫和,反應時間較短,活性高,催化劑用量少,無其他添加劑。
附圖說明
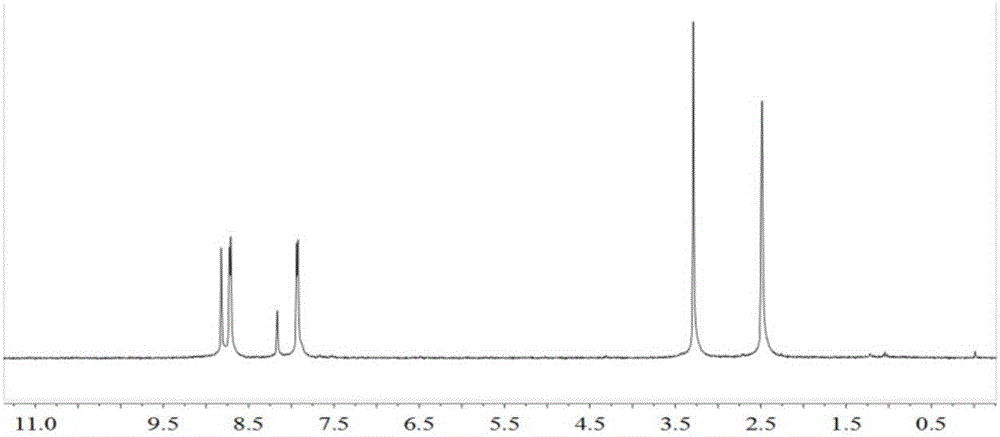
圖1為本發明實施例1氧化吡啶配體L的1HNMR;
圖2為本發明的氧化吡啶配體L的紅外譜圖;
圖3為本發明的Zn-MOF的紅外譜圖;
圖4為本發明的Zn-MOF的TGA譜圖;
圖5為本發明的Zn-MOF的單晶結構圖、一維鏈狀結構和手性二維面結構,其中,A為本發明的單晶結構圖,B為本發明一維鏈狀結構圖,C為本發明手性二維面結構圖;
圖6為本發明的Zn-MOF在催化Knoevenagel縮合反應后的對應PXRD譜圖。
具體實施方式
下面結合附圖及實施例對本發明作進一步說明。
實施例1:氧化吡啶配體L的制備
具體的制備步驟如下:
(1)在250mL單口瓶中,將3,5-二溴吡啶(3.2g,13.5mmol)溶于50mL THF,加入間氯過氧苯甲酸(6.88g,40mmol),室溫攪拌48小時,反應結束后加入飽和碳酸鈉水溶液30mL,靜置分層,水相用2×50mL CH2Cl2萃取,合并有機相并用無水Na2SO4干燥,硅膠柱層析(CH2Cl2)得淺白色固體1.53g,即為3,5-二溴吡啶-N-氧化物,產率44.1%。1H NMR(300MHz,DMSO,25℃,TMS):δ=8.60(s,2H,-C5H3NBr2),7.97(s,1H,-C5H3NBr2)。
(2)N2保護下,3,5-二溴吡啶-N-氧化物(2.53g,10.0mmol),4-吡啶硼酸(2.95g,24.0mmol),四三苯基磷合鈀(1.16g,1.0mmol),無水碳酸鉀(17.25g,125.0mmol),乙醇60mL,水60mL于三口瓶中,控溫75℃,攪拌48h。減壓蒸除溶劑,水洗,真空干燥,柱層析(CH2Cl2:CH3OH=20:1),得到白色粉末,即為氧化吡啶配體L。
對獲得的氧化吡啶配體L進行核磁共振氫譜及紅外譜圖的表征,其結果如分別如圖1、2所示。
實施例2:Zn-MOF的合成
在室溫下,將ZnBr2(2.2mg,0.01mmol)溶于甲醇(2mL)獲得ZnBr2的甲醇溶液,將L(2.4mg,0.01mmol)溶于CH2Cl2溶液(2mL)中,將甲醇溶液慢慢鋪在CH2Cl2溶液上,靜置三天后得到無色單晶。
通過紅外譜圖(IR),熱重分析譜圖(TGA)表征了該化合物,結果分別見圖3、圖4。
Zn-MOF單晶結構如圖5所示。
由圖5可以看出,Zn-MOF是單斜晶系,屬于P2(1)/n空間群。Zn-MOF中只存在一種Zn(II)中心,其處于(ZnN2Br2)的配位環境中,和它配位的氮原子是來自于配體L的端基吡啶,Zn-N鍵的長度分別是和其中,配體L中氧化吡啶的氧未參與配位,Zn(II)中心通過配體末端的吡啶氮原子相連,沿b軸方向伸展為一維螺旋鏈。一條鏈上氧化吡啶上的氧原子和另外一條鏈上氧化吡啶上的氫原子形成氫鍵,這些氫鍵將上述一維螺旋鏈進一步連接為波浪狀的手性二維面。Zn(II)和Zn(II)之間的最短的距離是具體晶體數據見表1
表1 Zn-MOF的晶體學數據
實驗例3:苯甲醛與丙二腈縮合反應生成
室溫下,在2mL玻璃瓶中加入苯甲醛(1mmol)與丙二腈(1.2mmol),攪拌5分鐘后,加入催化劑Zn-MOF(19mg,4%(mol),以苯甲醛的量為基準),繼續攪拌6h,用氣相色譜追蹤反應,反應結束后,快速離心,離心后的固體即為用于催化反應的催化劑Zn-MOF,回收催化劑,直接投入下一循環反應,按照上述條件,催化劑使用5個循環,反應液通過氣相色譜計算產率,催化效果如表2所示,回收的催化劑通過PXRD表征,如圖6所示。
表2 Zn-MOF催化Knoevenagel縮合反應5個循環的產率和TOF值
a:產率通過GC測定b:TOF=%yield(mmol of substrate/mmol of cat.h)
上述雖然結合附圖對本發明的具體實施方式進行了描述,但并非對發明保護范圍的限制,所屬領域技術人員應該明白,在本發明的技術方案的基礎上,本領域技術人員不需要付出創造性勞動即可做出的各種修改或變形仍在本發明的保護范圍內。
- 還沒有人留言評論。精彩留言會獲得點贊!