一種雷迪帕韋的制備方法及制備雷迪帕韋的中間體與流程
本發明涉及有機合成制藥的技術領域,特別涉及一種雷迪帕韋的制備方法及制備雷迪帕韋的中間體。
背景技術:
丙型肝炎是由丙肝病毒感染引起的,丙肝病毒為直徑約55~65nm球形顆粒,外有脂質囊膜和棘突結構,內有核心蛋白(core)和RNA組成的核衣殼。丙肝病毒的基因組含10個基因,表達產生10個結構蛋白(包含核心蛋白、包膜蛋白E1和E2、離子通道蛋白P7)和非結構蛋白(包含NS2、NS3、NS4A、NS4B、NS5A和NS5B)。其中NS5A是一種高度磷酸化的非結構蛋白,不具備酶催化活性,其磷酸化水平在HCV基因組的復制和翻譯過程中都起著調節的作用。NS5A上絲氨酸殘基產生兩種磷酸化程度不同的蛋白,即基礎磷酸化p56和高度磷酸化p58,均在HCV的生命周期中發揮重要作用。NS5A功能的重要性和多樣性使它成為抗HCV的重要靶點,其抑制劑的研究開發已經取得了非常引人矚目的成就。
Harvoni是索非布韋和雷迪帕韋的復方,臨床研究顯示,HCV患者接受這個藥物治療8周或12周,治愈率可達99%;復方中的雷迪帕韋是丙肝病毒NS5A抑制劑。
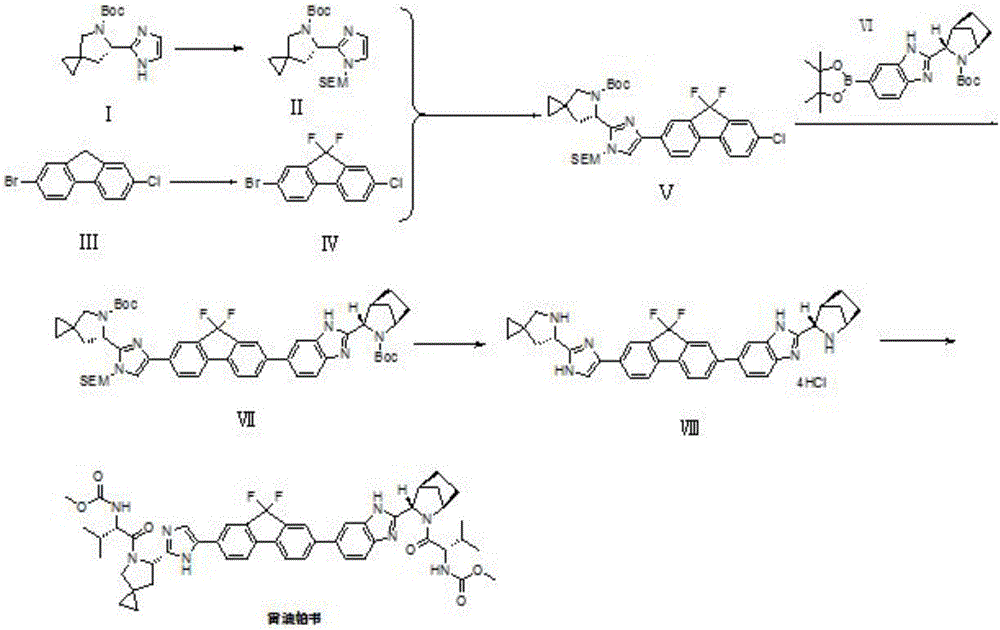
專利WO2013184702最早報道了雷迪帕韋,并且報道了由化合物XIII與化合物VI進行Suzuki偶聯反應來合成雷迪帕韋的方法,合成路線如下式所示。其中涉及到使用昂貴的N-氟代雙苯磺酰胺作為氟代試劑合成化合物X,化合物X再經過格式反應、酯化反應、環化反應、Suzuki偶合反應和脫保護反應共五步反應得到制備雷迪帕韋的關鍵中間體化合物VIII,該方法反應步驟多,制備過程過于復雜。
技術實現要素:
有鑒于此,本發明目的在于提供一種工藝簡便、步驟少的雷迪帕韋的制備方法。
為了實現上述發明目的,本發明提供以下技術方案:
本發明提供了一種雷迪帕韋關鍵中間體的制備方法,包括以下步驟:
(1)在堿性物質的作用下,將具有式I所示結構的化合物和2-(三甲基硅烷基)乙氧甲基氯在非質子有機溶劑中進行咪唑環保護反應,得到具有式Ⅱ所示結構的化合物;
(2)在有機胺鹽的作用下,將2-溴-7-氯-9H-芴與N-氟代雙苯磺酰胺在醚類溶劑中進行氟代反應,得到2-溴-7-氯-9,9-二氟-9H-芴;
(3)在堿性物質、鈀催化劑和有機磷配體的作用下,將所述具有式Ⅱ所示結構的化合物和所述2-溴-7-氯-9,9-二氟-9H-芴在極性溶劑中進行碳-氫活化偶聯反應,得到具有式Ⅴ所示結構的化合物;
(4)在堿性物質、鈀催化劑和有機磷配體的作用下,將所述具有式Ⅴ所示結構的化合物和具有式Ⅵ所示結構的化合物在極性溶劑中進行進行Suzuki偶聯反應,得到具有式Ⅶ所示結構的化合物;
(5)將所述具有式Ⅶ所示結構的化合物和酸性物質在非質子極性溶劑中進行脫保護基反應,得到雷迪帕韋關鍵中間體,具有式Ⅷ所示結構;
所述步驟(1)和步驟(2)之間沒有時間順序限制。
優選的,所述步驟(1)中的堿性物質為氫化鈉、叔丁醇鉀、叔丁醇鈉、雙(三甲基硅烷基)氨基鋰、雙(三甲基硅烷基)氨基鈉和雙(三甲基硅烷基)氨基鉀中的一種或幾種的混合物;
優選的,所述步驟(1)中的非質子有機溶劑為四氫呋喃、2-甲基四氫呋喃、乙二醇二甲醚和N,N-二甲基甲酰胺中的一種或幾種的混合物。
優選的,所述步驟(2)中的有機胺鹽為雙(三甲基硅烷基)氨基鉀、雙(三甲基硅烷基)氨基鋰、雙(三甲基硅烷基)氨基鈉和二異丙基氨基鋰中的一種或幾種的混合物;
優選的,所述步驟(2)中的醚類溶劑為四氫呋喃、2-甲基四氫呋喃、異丙醚和甲基叔丁基醚中的一種或幾種的混合物。
優選的,所述步驟(3)中的堿性物質為碳酸鉀、叔丁醇鉀、碳酸銫和醋酸銫中的一種或幾種的混合物;
優選的,所述步驟(3)中的鈀催化劑為Pd(OAc)2、Pd2(dba)3、Pd(PPh3)4和Pd(PPh3)2Cl2中的一種或幾種的混合物;
優選的,所述步驟(3)中的有機磷配體為2-二環己基膦-2′,6′-二甲氧基-聯苯、正丁基二(1-金剛烷基)膦和2-二環己基磷-2,4,6-三異丙基聯苯中的一種或幾種的混合物;
優選的,所述步驟(4)中的堿性物質為碳酸鉀、碳酸鈉、碳酸銫和碳酸氫鈉中的一種或幾種的混合物;
優選的,所述步驟(4)中的鈀催化劑為Pd(OAc)2、Pd(dppf)Cl2、Pd2(dba)3和Pd(PPh3)4中的一種或幾種的混合物。
優選的,所述步驟(4)中的有機磷配體為2-二環己基膦-2′,6′-二甲氧基-聯苯、4,5-雙二苯基膦-9,9-二甲基氧雜蒽和2-二環己基磷-2,4,6-三異丙基聯苯中的一種或幾種的混合物。
優選的,所述步驟(5)中的酸性物質為鹽酸、氫溴酸、甲磺酸、對甲苯磺酸和三氟醋酸中的一種或幾種的混合物。
優選的,所述步驟(4)為:當步驟(3)和步驟(4)中使用的堿和有機磷配體種類一致時,將所述步驟(3)得到的反應液中與具有式Ⅵ所示結構的化合物和鈀催化劑混合,進行Suzuki偶聯反應,得到具有式Ⅶ所示結構的化合物。
本發明提供了一種利用上述方案所述制備方法制備的關鍵中間體制備雷迪帕韋的方法,包括以下步驟:
在堿性物質和縮合劑的作用下,將所述雷迪帕韋關鍵中間體與Moc-L-纈氨酸在極性溶劑中進行縮合反應,得到雷迪帕韋。
本發明提供了用于制備雷迪帕韋的中間體化合物,具有式XV所示結構:
式XV中,R為H、
本發明提供了一種雷迪帕韋的制備方法,以具有式Ⅰ所示結構的化合物和2-溴-7-氯-9H-芴為起始物,分別制得具有式II所示結構的化合物和2-溴-7-氯-9,9-二氟-9H-芴,再經過碳-氫活化偶聯反應、Suzuki偶聯反應和脫保護基反應,即可得到合成雷迪帕韋的關鍵中間體—具有式Ⅷ所示結構的化合物,具有式Ⅷ所示結構的化合物再與Moc-L-纈氨酸進行縮合反應,即可得到雷迪帕韋。本發明提供的雷迪帕韋的制備方法簡單、步驟少,關鍵化合物2-溴-7-氯-9,9-二氟-9H-芴(式Ⅳ)僅需經過碳-氫活化偶聯反應、Suzuki偶聯反應和去保護基反應,三步即可得到制備雷迪帕韋的關鍵中間體VIII,提高了氟化合物的利用率,降低了制備成本,實驗結果表明,本發明的合成路線中Suzuki偶聯反應的收率可以達到80%以上,去保護基反應的收率可以達到95%,氟化物的利用率可以達到50%以上。
附圖說明
圖1為本發明實施例合成雷迪帕韋的路線圖。
具體實施方式
本發明提供了一種雷迪帕韋關鍵中間體的制備方法,包括以下步驟:
(1)在堿性物質的作用下,將具有式I所示結構的化合物和2-(三甲基硅烷基)乙氧甲基氯在非質子有機溶劑中進行咪唑環保護反應,得到具有式Ⅱ所示結構的化合物;
(2)在有機胺鹽的作用下,將2-溴-7-氯-9H-芴與N-氟代雙苯磺酰胺在醚類溶劑中進行氟代反應,得到2-溴-7-氯-9,9-二氟-9H-芴;
(3)在堿性物質、鈀催化劑和有機磷配體的作用下,將所述具有式Ⅱ所示結構的化合物和所述2-溴-7-氯-9,9-二氟-9H-芴在極性溶劑中進行碳-氫活化偶聯反應,得到具有式Ⅴ所示結構的化合物;
(4)在堿性物質、鈀催化劑和有機磷配體的作用下,將所述具有式Ⅴ所示結構的化合物和具有式Ⅵ所示結構的化合物在極性溶劑中進行Suzuki偶聯反應,得到具有式Ⅶ所示結構的化合物;
(5)將所述具有式Ⅶ所示結構的化合物和酸性物質在非質子極性溶劑中進行脫保護基反應,得到雷迪帕韋關鍵中間體,具有式Ⅷ所示結構;
所述步驟(1)和步驟(2)之間沒有時間順序限制。
本發明在堿性物質的作用下,將具有式I所示結構的化合物((S)-6-(1H-咪唑-2-基)-5-螺雜環[2.4]庚烷-5-羧酸叔丁酯)和2-(三甲基硅烷基)乙氧甲基氯在非質子有機溶劑中進行咪唑環保護反應,得到具有式Ⅱ所示結構的化合物((S)-6-[1-((2-(三甲基硅基)乙氧基)甲基-1H-咪唑-2-基]-5-螺雜環[2.4]庚烷-5- 羧酸叔丁酯)。在本發明中,所述咪唑環保護反應用的堿性物質優選氫化鈉、叔丁醇鉀、叔丁醇鈉、雙(三甲基硅烷基)氨基鋰、雙(三甲基硅烷基)氨基鈉和雙(三甲基硅烷基)氨基鉀中的一種或幾種的混合物;所述咪唑環保護反應用的非質子有機溶劑優選為四氫呋喃、2-甲基四氫呋喃、乙二醇二甲醚和N,N-二甲基甲酰胺中的一種或幾種的混合物。
在本發明中,所述具有式I所示結構的化合物、堿性物質和2-(三甲基硅烷基)乙氧甲基氯的物質的量比優選為1:1~4:1~3,更優選為1:2~3:1.5~2.5;所述具有式I所示結構的化合物的質量與非質子有機溶劑的體積比優選為1g:15~25ml,更優選為1g:20ml。
在本發明中,所述咪唑環保護反應的溫度優選為室溫;所述咪唑環保護反應的時間優選為1.5~3h,更優選為2h。本發明優選在攪拌的條件下進行咪唑環保護反應;所述攪拌的速率優選為300~500r/min,更優選為350~450r/min。
在本發明的部分具體實施例中,可以將具有式I所示結構的化合物和非質子有機溶劑混合,得到具有式I所示結構的化合物溶液;在0~5℃條件下將堿性物質和具有式I所示結構的化合物溶液混合,攪拌0.5~1h,得到第一混合溶液;將2-(三甲基硅烷基)乙氧甲基氯滴加至第一混合溶液中,控制滴加過程中第一混合液的溫度為0~5℃,得到第二混合溶液;將第二混合溶液自然升溫至室溫進行咪唑環保護反應,所述反應時間自升溫至室溫時開始計算。在本發明中,滴加過程中控制混合溶液的溫度優選為0~5℃;所述滴加的速度優選為1~5滴/秒,更優選為2~3滴/秒。
所述咪唑環保護反應完成后,本發明優選將得到咪唑環保護反應液進行后處理,得到具有式Ⅱ所示結構的化合物。在本發明中,咪唑環保護反應液的后處理過程優選包括:將咪唑環保護反應淬滅,依次對得到的反應液進行萃取、洗滌、濃縮和純化,得到式Ⅱ所示結構的化合物。
在本發明中,所述淬滅反應用淬滅劑優選為水;本發明優選使用薄層色譜法檢測反應歷程,薄層色譜法檢測顯示反應結束時,將反應淬滅。在本發明中,所述萃取用萃取劑優選為乙酸乙酯;所述萃取的次數優選為2~5次,更優選為3~4次;本發明將所述萃取得到有機相洗滌,所述洗滌的洗滌劑優選為飽和氯化鈉溶液;所述洗滌的次數優選為1~3次。在本發明中,所述濃縮優選為減壓濃縮;本發明優選將所述洗滌產物濃縮至干。在本發明中,所述純化優選為硅膠柱純化;所述硅膠柱純化用展開劑優選為乙酸乙酯和石油醚的混合物;所述乙酸乙酯和石油醚的混合物中乙酸乙酯和石油醚的體積比優選為1:1~5,更優選為1:2~4。
本發明對式Ⅰ所示化合物的來源沒有特殊要求,使用市售商品或使用本領域常規方法進行合成均可。
本發明在有機胺鹽的作用下,將2-溴-7-氯-9H-芴(式Ⅲ)與N-氟代雙苯磺酰胺在醚類溶劑中進行氟代反應,得到2-溴-7-氯-9,9-二氟-9H-芴(式Ⅳ)。在本發明中,所述氟代反應用有機胺鹽優選為雙(三甲基硅烷基)氨基鉀、雙(三甲基硅烷基)氨基鋰、雙(三甲基硅烷基)氨基鈉和二異丙基氨基鋰中的一種或幾種的混合物;所述氟代反應用醚類溶劑優選為四氫呋喃、2-甲基四氫呋喃、異丙醚和甲基叔丁基醚中的一種或幾種的混合物。
在本發明中,所述2-溴-7-氯-9H-芴、有機胺鹽與N-氟代雙苯磺酰胺的物質的量比優選為1:2.5~4.5:2~5,更優選為1:3.0~4.0:2.5~3.0;所述2-溴-7-氯-9H-芴的質量與醚類溶劑的體積比優選為1g:20~25ml,更優選為1g:21~23ml。
本發明對2-溴-7-氯-9H-芴的來源沒有特殊要求,使用使用本領域常規方法進行合成或直接使用市售的商品均可。
在本發明中,所述氟代反應的溫度優選為-60~-100℃,更優選為-70~-80℃;所述氟代反應的時間優選為1.5~3h,更優選為2h;本發明優選在惰性氣體保護下進行氟代反應。
在本發明的部分具體實施例中,可以將具有式Ⅲ所示結構的化合物、N-氟代雙苯磺酰胺和第一部分醚類溶劑混合,得到混合溶液;將堿性化合物溶解于第二部分醚類溶劑中,得到堿溶液;將所述堿溶液滴加到混合溶液中,進行氟代反應。在本發明中,滴加過程中控制混合溶液的溫度為-70~-80℃;所述滴加的速度優選為1~5滴/秒,更優選為2~3滴/秒;所述第一部分醚類溶劑和第二部分醚類溶劑的總體積符合上述比例,在此不再贅述;所述第一部分醚類溶劑和第二部分醚類溶劑的體積比優選為1:0.9~1.2,更優選為1:1~1.1;本發明的反應時間自堿溶液滴加完畢開始計算。
所述氟代反應完成后,本發明優選將得到的氟代反應液進行后處理,得到2-溴-7-氯-9,9-二氟-9H-芴。在本發明中,所述后處理優選包括以下步驟:
將氟代反應液自然升溫至-10~-20℃,向反應液中加入水后濃縮,得到濃縮物;
將所述濃縮物過濾,得到第一濾餅,將所述第一濾餅與甲醇混合,在30~50℃下攪拌0.5~1h,過濾得到第二濾餅;
將所述第二濾餅與甲苯混合,在80~95℃下攪拌1~1.5h,趁熱過濾,濾液旋干得到第三濾餅;
將所述第三濾餅與異丙醇混合,在0~5℃下攪拌1~1.5h,過濾,得到第四濾餅;
將所述第四濾餅用異丙醇洗滌后干燥,得到2-溴-7-氯-9,9-二氟-9H-芴。
在本發明中,所述水的加入量優選為反應液體積的0.5~2.0倍,更優選為1.0~1.5倍;所述濃縮優選為減壓濃縮;本發明優選將反應液濃縮至原體積的1/5~1/3;
在本發明中,所述甲醇的體積和第一濾餅的重量比優選為2~5ml:1g,更優選為3~4ml:1g;所述攪拌的轉速優選為300~500r/min,更優選為350~450r/min;
在本發明中,所述甲苯的體積和第二濾餅的重量比優選為2~5ml:1g,更優選為3~4ml:1g;
在本發明中,所述異丙醇的體積和第三濾餅的重量比優選為4~7ml:1g,更優選為5~6ml:1g;
在本發明中,所述使用異丙醇洗滌的次數優選為2~5次,更優選為3~4次;所述干燥的溫度優選為40~60℃,更優選為50℃,干燥至恒重即可;
在上述后處理過程中,所述各步驟中攪拌的轉速獨立的優選為300~500r/min,更優選為350~450r/min。
得到具有式Ⅱ所示結構的化合物和2-溴-7-氯-9,9-二氟-9H-芴后,本發明在堿性物質、鈀催化劑和有機磷配體的作用下,將所述具有式Ⅱ所示結構的化合物和所述2-溴-7-氯-9,9-二氟-9H-芴在極性溶劑中進行碳-氫活化偶聯反應,得到具有式Ⅴ所示結構的化合物((S)-6-(4-(7-氯-9,9-二氟-9H-芴-2-基)-1-[(2-(三甲基硅基)乙氧基)甲基-1H-咪唑-2-基]-5-螺雜環[2.4]庚烷-5-羧酸叔丁酯)。
在本發明中,所述碳-氫活化偶聯反應用堿性物質優選為碳酸鉀、叔丁醇鉀、碳酸銫和醋酸銫中的一種或幾種的混合物;所述碳-氫活化偶聯反應用鈀催化劑優選為Pd(OAc)2、Pd2(dba)3、Pd(PPh3)4和Pd(PPh3)2Cl2中的一種或幾種的混合物;所述碳-氫活化偶聯反應用有機磷配體優選為2-二環己基膦-2′,6′-二甲氧基-聯苯(SPhos)、正丁基二(1-金剛烷基)膦(P(n-Bu)Ad2)和2-二環己基磷-2,4,6-三異丙基聯苯(XPhos)中的一種或幾種的混合物;所述碳-氫活化偶聯反應用極性溶劑的極性值優選為4.5~7.5,更優選為5.0~7.0;所述極性溶劑具體的優選為二氧六環、N,N-二甲基乙酰胺、N-甲基吡咯烷酮和乙二醇二甲醚中的一種或幾種的混合物。
在本發明中,所述具有式Ⅱ所示結構的化合物、2-溴-7-氯-9,9-二氟-9H-芴、堿性物質、鈀催化劑和有機磷配體的物質的量比優選為1:1~1.5:2~3:0.005~0.1:0.01~0.2,和更優選為1:1.2~1.3:2.3~2.5:0.01~0.05:0.05~0.15;所述具有式Ⅱ所示結構的化合物的質量與極性溶劑的體積比優選為1:10~20ml,更優選為1:13~16ml。
在本發明中,所述碳-氫活化偶聯反應的溫度優選為110~130℃,更優選為115~125℃,最優選為120℃;所述碳-氫活化偶聯反應的時間優選為11~13h,最優選為12h。
所述碳-氫活化偶聯反應后,本發明優選將得到的碳-氫活化偶聯反應液過濾,向得到的濾液中加水淬滅反應,再依次對所述淬滅后的濾液進行萃取、洗滌和濃縮,得到具有式Ⅴ所示結構的化合物的粗產品,粗產品可直接應用于下一步的制備。在本發明中,所述萃取用萃取劑優選為乙酸乙酯;所述萃取劑與濾液的體積比優選為1.5~3.5:1,更優選為2~3:1;所述萃取的次數優選為2~5次,更優選為3~4次。本發明優選將萃取得到的有機相合并,進行洗滌;所述洗滌的洗滌劑優選為飽和氯化鈉溶液;所述洗滌的次數優選為1~3次。在本發明中,所述濃縮優選為減壓濃縮;本發明對減壓濃縮的具體操作方法沒有特殊要求,使用本領域常規的減壓濃縮方法即可,本發明優選濃縮至干,得到粗產品。
得到具有式Ⅴ所示結構的化合物后,本發明在堿性物質、鈀催化劑和有機磷配體的作用下,將所述具有式Ⅴ所示結構的化合物和具有式Ⅵ所示結構的化合物((1R,3S,4S)-3-[6-(4,4,5,5-四甲基-1,3,2-二氧雜環戊硼烷-2-基)-1H-苯并咪唑-2-基]-2-氮雜雙環[2.2.1]庚烷-2-羧酸叔丁酯)在極性溶劑中進行Suzuki偶聯反應,得到具有式Ⅶ所示結構的化合物((S)-6-(4-(7-(2-((1R,3S,4S)-2-(叔丁氧羰基)-2-氮雜雙環[2.2.1]庚烷-3-基)-1H-苯并[d]咪唑-6-劑)-9,9-二氟-9H-芴-2-基)-1-[(2-(三甲基硅基)乙氧基)甲基-1H-咪唑-2-基]-5-螺雜環[2.4]庚烷-5-羧酸叔丁酯)。在本發明中,所述Suzuki偶聯反應用堿性物質優選為碳酸鉀、碳酸鈉、碳酸銫和碳酸氫鈉中的一種或幾種的混合物;所述Suzuki偶聯反應用鈀催化劑不能為Pd(PPh3)2Cl2,其余和步驟(3)中使用的鈀催化劑種類相同,且還可以為Pd(dppf)Cl2;所述Suzuki偶聯反應用有機磷配體優選為2-二環己基膦-2′,6′-二甲氧基-聯苯(SPhos)、4,5-雙二苯基膦-9,9-二甲基氧雜蒽(XantePhos)和2-二環己基磷-2,4,6-三異丙基聯苯(XPhos)中的一種或幾種的混合物;所述Suzuki偶聯反應用極性溶劑的極性值優選為4.5~7.2,更優選為5~6.5;所述極性溶劑具體的優選為二氧六環、N,N-二甲基乙酰胺、N-甲基吡咯烷酮和乙二醇二甲醚中的一種或幾種的混合物。
在本發明中,所述具有式Ⅴ所示結構的化合物、具有式Ⅵ所示結構的化合物、堿性物質、鈀催化劑和有機磷配體的物質的量比優選為1:1~1.5:2~3:0.005~0.1:0.001~0.2,更優選為1:1.2~1.3:2.3~2.5:0.01~0.05:0.005~0.1;所述具有式Ⅴ所示結構的化合物的質量和極性溶劑的體積優選為1g:20~30ml,更優選為1g:25ml。
本發明對具有式Ⅵ所示結構的化合物的來源沒有特殊要求,使用本領域方法進行合成或直接使用市售的商品均可。
在本發明中,所述Suzuki偶聯反應的溫度優選為100~110℃,更優選為103~105℃;所述Suzuki偶聯反應的時間優選為6~8h,更優選為7h。
本發明還提供了一種通過“一鍋法”實現碳-氫活化偶聯反應和Suzuki偶聯反應的方法,即當步驟(3)和步驟(4)中使用的堿性物質和有機磷配體的種類一致時,所述步驟(3)的反應完成后,將步驟(3)的反應液與具有式Ⅵ所示結構的化合物和鈀催化劑混合進行Suzuki偶聯反應;步驟(3)中得到的具有式Ⅴ所示結構的化合物無需進行分離和純化處理;此處所用的鈀催化劑與上述方案步驟(4)中的鈀催化劑一致,在此不再贅述。
本發明提供的制備方法通過“一鍋法”來實現具有式Ⅶ所示結構的化合物的合成,將步驟(3)和步驟(4)綜合到一個反應器中,無需進行中間產物的分離和純化,進一步簡化了制備步驟,降低了生產成本。
所述Suzuki偶聯反應完成后,本發明優選將反應淬滅,依次對反應液進行萃取、洗滌、濃縮和純化,得到式Ⅶ所示結構的化合物。在本發明中,所述萃取、洗滌、濃縮和純化的方法與分離咪唑環保護反應產物時的方法一致,在此不再贅述。
得到具有式Ⅶ所示結構的化合物后,本發明將所述具有式Ⅶ所示結構的化合物和酸性物質在非質子極性溶劑中進行脫保護基反應,得到雷迪帕韋關鍵中間體,具有式Ⅷ所示結構。在本發明中,所述酸性物質優選為鹽酸、氫溴酸、甲磺酸、對甲苯磺酸和三氟醋酸中的一種或幾種的混合物;所述鹽酸優選為質量濃度為36~38%的濃鹽酸;所述氫溴酸優選為質量濃度為47.5%的濃氫溴酸;所述甲磺酸優選為純度大于98%的甲磺酸;所述對甲苯磺酸優選為純度大于96%的對甲苯磺酸;所述三氟醋酸優選為純度大于99%的濃三氟醋酸;所述脫保護基反應用非質子極性溶劑的極性值優選為4.5~7.2,更優選為5~6.5;所述非質子極性溶劑具體的優選為乙二醇二甲醚、丙酮和乙腈中的一種或幾種的混合物。
在本發明中,所述具有式Ⅶ所示結構的化合物和酸性物質的摩爾比優選為1:4.0~6.0,更優選為1:4.3~5.0;所述具有式Ⅶ所示結構的化合物的質量和非質子極性溶劑的體積比優選為1g:15~25ml,更優選為1g:20ml。
在本發明中,所述脫保護基反應的溫度優選為70~90℃,更優選為75~85℃,最優選為80℃;所述脫保護基反應的時間優選為5~7h,更優選為5.5~6.5h,最優選為6h;本發明優選在加熱回流的條件下進行所述脫保護基反應。
在本發明的部分具體實施例中,可以將具有式Ⅶ所示結構的化合物和非質子極性溶劑混合,得到具有式Ⅶ所示結構的化合物的溶液;將酸性物質滴加至具有式Ⅶ所示結構的化合物的溶液中進行脫保護劑反應,得到具有式VIII所示結構的化合物。在本發明中,所述滴加速度優選為1~5滴/秒,更優選為2~3滴/秒;本發明的反應時間自酸性物質滴加完畢開始計算。
所述脫保護基反應完成后,本發明優選將脫保護基反應液進行后處理,得到雷迪帕韋關鍵中間體。在本發明中,所述脫保護基反應液后處理的方法優選具體為:將反應液過濾,得到濾餅;將所述濾餅用乙酸乙酯洗滌后濃縮去除殘留溶劑。
本發明提供了一種利用上述方案所述制備方法制備的關鍵中間體制備雷迪帕韋的方法,包括以下步驟:
在堿性物質和縮合劑的作用下,將所述雷迪帕韋關鍵中間體與Moc-L-纈氨酸在極性溶劑中進行縮合反應,得到雷迪帕韋。
在本發明中,所述縮合反應用堿性物質優選為有機堿;更優選為N-甲基嗎啉、三乙胺和二異丙基乙胺中的一種或幾種的混合物;所述縮合劑優選為1-乙基-(3-二甲基氨基丙基)碳酰二亞胺鹽酸鹽(EDC.HCl)和1-羥基苯并三唑(HOBt)或2-(7-氧化苯并三氮唑)-N,N,N',N'-四甲基脲六氟磷酸酯(HATU)中的一種或幾種的混合物;所述縮合反應用極性溶劑的極性值優選為4.5~7.2,更優選為5.0~6.5;所述極性溶劑具體的優選為N,N-二甲基甲酰胺、N,N-二甲基乙酰胺、N-甲基吡咯烷酮和二氯甲烷中的一種或幾種的混合物。
在本發明中,所述雷迪帕韋關鍵中間體、縮合劑、堿性物質和Moc-L-纈氨酸的物質的量比優選為1:3~8:8~14:2~4,更優選為1:4~6:9~11:2.5~3.5;所述具有式Ⅷ所示結構的化合物的質量和極性溶劑的體積比優選為1g:8~12ml,更優選為1g:9~11ml,最優選為1g:10ml。
在本發明中,所述縮合反應的溫度優選為0~40℃,更優選為10~30℃;所述縮合反應的時間優選為2~3h,更優選為2.5~2.8h。
在本發明的部分具體實施例中,可以將酸性物質、Moc-L-纈氨酸和極性溶劑混合并在0~40℃條件下攪拌0.5~1h,得到酸性物質和Moc-L-纈氨酸的混合溶液;將所述混合溶液溫度降至0~5℃,向所述混合溶液中加入具有式Ⅷ所示結構的化合物,得到反應原液;將有機堿滴加至所述反應原液中,滴加完畢后將溶液溫度升至2~30℃進行縮合反應,得到雷迪帕韋。在本發明中,所述滴加的速度優選為1~5滴/秒,更優選為2~3滴/秒;所述縮合反應的時間自升溫至所需溫度時開始計算;本發明步驟(6)中使用的有機堿均為液態物質,可以直接滴加,無需使用溶劑進行溶解。
所述縮合反應完成后,本發明優選將縮合反應產物分離,得到雷迪帕韋。在本發明中,所述分離縮合反應產物優選具體為:向反應液中加入乙酸乙酯和水,攪拌0.5~1h后靜置分層,得到水相和第一有機相;將所述水相用乙酸乙酯萃取,得到第二有機相;將所述第二有機相和第一有機相合并后與吸水性物質混合,再攪拌1~1.5h后過濾,得到濾液;將所述濾液濃縮,得到濃縮物;將所述濃縮物與丙酮混合,在5~15℃下攪拌12~24h后過濾和干燥,得到雷迪帕韋粗產品;將所述雷迪帕韋粗產品與乙酸乙酯混合后過濾、濃縮至干,得到濃縮固體;將所述濃縮固體用丙酮溶解至澄清后,攪拌10~12h,析出固體后過濾和干燥,得到雷迪帕韋純品。
在本發明中,所述反應液中加入的乙酸乙酯和反應液的體積比優選為1.8~2.2:1,更優選為2:1;所述反應液中加入的乙酸乙酯和水的體積比優選為2~3:1,更優選為2.3~2.5:1;
在本發明中,所述萃取用乙酸乙酯與水相的體積比優選為1.5~3.5:1,更優選為2~3:1;所述萃取的次數優選為2~3次;
在本發明中,所述吸水性物質優選為無水硫酸鈉和/或硅膠;所述吸水性物質的加入量優選為0.05~0.2g/ml,更優選為0.1~0.15g/ml;
在本發明中,所述濃縮濾液的溫度優選為70~100℃,更優選為80~90℃;所述濃縮濾液的時間優選為1~3h,更優選為2h;本發明優選將濾液濃縮至原體積的1/5~1/3,更優選為濃縮至原體積的1/4;
在本發明中,所述丙酮與濃縮物的體積比優選為4~5:1,更優選為4.5:1;所述雷迪帕韋粗產品的干燥溫度優選為70~90℃,更優選為75~85℃,干燥時間優選為1~3h,更優選為1.5~2.5h;
在本發明中,所述雷迪帕韋粗產品與乙酸乙酯的質量比優選為1:3~3.5,更優選為1:3.1~3.3;
在本發明中,所述濃縮得到濃縮固體的步驟中的濃縮的溫度優選為70~100℃,更優選為80~90℃,本發明此處對濃縮時間沒有特殊要求,濃縮至干即可;
在本發明中,所述濃縮固體與丙酮的質量比優選為1:10~12,更優選為1:11;
在本發明中,所述雷迪帕韋粗純品的干燥溫度和時間與雷迪帕韋粗產品的干燥溫度和時間一致,在此不再贅述。
本發明的制備過程中合成了用于制備雷迪帕韋的新的中間體化合物,具有式XV所示結構:
式XV中,R為H、該中間體化合物用于合成雷迪帕韋,能夠減少合成步驟,提高合成效率。
下面結合實施例對本發明提供的雷迪帕韋的制備方法進行詳細的說明,但是不能把它們理解為對本發明保護范圍的限定。
實施例1
(1)在250mL的四口瓶中加入5.0g具有式Ⅰ所示結構的化合物和100mL四氫呋喃,降溫至0℃,加入質量分數為60%的分散于石蠟油的氫化鈉912mg,溫度控制在0℃;氫化鈉加入結束后,在0℃下攪拌反應30分鐘。滴加3.3g 2-(三甲基硅烷基)乙氧甲基氯,滴加過程溫度控制在0℃,滴加結束后自然升溫至室溫,攪拌反應2小時;反應完成后取樣,TLC(PE:EA=1:1)檢測顯示反應結束;
對產物進行分離提純:緩慢滴加100mL水淬滅反應,用150mL乙酸乙酯萃取三次,合并有機相用40mL飽和食鹽水洗滌一次,減壓濃縮至干,濃縮物用快速硅膠柱純化,展開劑為乙酸乙酯和石油醚混合物(PE:EA=10:1到PE:EA=3:1),收集含有產品的組分,濃縮得具有式Ⅱ所示結構的化合物5.2g,收率69.6%,純度97%;
使用核磁共振對產物結構進行檢測,可得1HNMR譜圖數據為:1HNMR(400MHz CDCl3)δ6.99(s,1H),6.90(s,1H),5.78-5.73(m,0.5H),5.44–5.35(m,0.5H),5.20–5.13(m,1H),5.10–5.02(m,1H),3.80-3.64(m,1H),3.53–3.45(m,2H),3.33(d,1H,J=10Hz),2.33–2.23(m,1H),2.10-1.95(m,1H),1.40(s,4.5H),1.22(s,4.5H),1.00-0.83(m,2H),0.78–0.66(m,1H),0.64–0.58(m,2H),0.57–0.52(m,1H);
根據核磁共振結果可知,所得產物確實為具有式Ⅱ所示結構的化合物;
(2)在2.0L反應瓶中加入60g 2-溴-7-氯-9H-芴,186g N-氟代雙苯磺酰胺和660mL四氫呋喃,在氮氣保護下,反應液降溫至-78℃,緩慢滴加690mL1.0mol/L的雙(三甲基硅烷基)氨基鋰的四氫呋喃溶液,滴加時溫度控制在-70℃,滴加結束后,在-70℃下攪拌反應2小時;取樣送LC-MS檢測顯示反應結束;
對產物進行分離提純:將反應液自然升溫至-20℃,滴加108mL水至澄清,減壓濃縮至大量固體析出,分三批加入840mL水帶蒸四氫呋喃,冷卻至20℃,過濾;濾餅加入180g甲醇中,升溫至40℃攪拌30分鐘,降溫至20℃攪拌30分鐘,過濾;濾餅加入192g甲苯中,升溫至90℃攪拌1小時;布氏漏斗中鋪少量硅膠,趁熱過濾;濾餅用熱甲苯洗滌兩次;減壓濃縮大部分甲苯,滴加300g異丙醇,降溫至5℃攪拌1小時,過濾,濾餅用異丙醇洗滌兩次,濾餅在50℃下鼓風干燥得43.8g2-溴-7-氯-9,9-二氟-9H-芴;收率63.9%,純度96.6%;
使用核磁共振對產物結構進行檢測,可得1HNMR譜圖數據為:1H NMR(400MHz,CDCl3),δ:7.80–7.70(m,1H),7.70–7.56(m,2H),7.46(q,J=8.4Hz,2H),7.50–7.36(m,1H);
根據核磁共振結果可知,所得產物確實為具有式Ⅳ所示結構的化合物;
(3)在氮氣保護下,向250mL單口瓶中加入6.2g具有式Ⅱ所示結構的化合物,4.1g 2-溴-7-氯-9,9-二氟-9H-芴,3.6g碳酸鉀,147mg Pd(OAc)2,550mg SPhos和90mL N,N-二甲基乙酰胺,加完后,反應體系升溫至120℃攪拌反應12小時;取樣TLC(PE:EA=3:1)檢測顯示反應結束;
將產物進行分離提純:向反應液中加少量硅藻土,過濾除去黑色不溶物;向濾液中,加150mL水淬滅反應,用300mL乙酸乙酯萃取兩次;合并有機相,用飽和食鹽水洗滌兩次,減壓濃縮干;得到具有式Ⅴ所示結構的化合物的粗品,粗產品可直接用于下一步反應。
取樣品,采用硅膠柱方法純化,將得到的純品進行質譜和核磁共振檢測;所得數據為:MS=628[M++1];1HNMR(400MHz CDCl3)δ7.80–7.70(m,1H),7.63–7.54(m,3H),7.52–7.48(m,1H),7.45–7.42(m,1H),7.17–7.11(m,1H),5.70-5.60(m,0.5H),5.33–5.28(m,0.5H),5.20–5.13(m,2H),3.92-3.64(m,1H),3.53–3.45(m,2H),3.40–3.33(m,1H),2.12–2.07(m,1H),1.95-1.85(m,1H),1.40(s,4.5H),1.22(s,4.5H),0.97-0.92(m,2H),0.87–0.81(m,2H),0.68–0.62(m,1H),0.57–0.52(m,1H),0.00(s,9H);
根據檢測結果可知,所得產物確實為具有式Ⅴ所示結構的化合物;
(4)在250mL單口瓶中加入4.0g具有式Ⅴ所示結構的化合物,3.4g具有式Ⅵ所示結構的化合物,1.8g碳酸鉀,143mg Pd(dppf)Cl2,526mg SPhos,80mL 1.4-二氧六環和20mL水;反應體系用氮氣置換三次,升溫至100℃回流7小時;取樣送LC-MS檢測顯示反應結束;
將產物分離提純:加少量硅藻土和50mL水淬滅反應,加100mL乙酸乙酯萃取三次,合并有機相,用飽和食鹽水洗滌兩次,減壓濃縮干;濃縮物用快速硅膠柱純化,展開劑為乙酸乙酯和石油醚混合物(PE:EA=10:1到PE:EA=3:1),收集含有產品的組分,濃縮、干燥得具有式Ⅶ所示結構的化合物4.7g,純度98%,收率為82%;
使用核磁共振對產物結構進行質譜和核磁共振檢測,所得譜圖數據為:MS=453[M/2+1];1HNMR(400MHz CDCl3)δ10.88–10.62(m,1H),7.90(s,1H),7.80–7.70(m,2H),7.63–7.54(m,2H),7.52–7.48(m,3H),7.18–7.10(m,1H),5.70-5.60(m,0.5H),5.33–5.28(m,0.5H),5.20–5.13(m,2H),4.58(s,1H),4.18(s,1H),3.82-3.64(m,1H),3.53–3.45(m,2H),3.40–3.33(m,1H),2.12–2.07(m,2H),1.78-1.70(m,3H),1.52(s,9H),1.45-1.38(m,4H),1.24(s,9H),0.97-0.92(m,2H),0.87–0.81(m,4H),0.68–0.62(m,2H),0.00(s,9H).
根據檢測結果可知,所得產物確實為具有式Ⅶ所示結構的化合物。
(5)在250mL單口瓶中加入2.5g具有式Ⅶ所示結構的化合物和50mL DME,滴加1.3g濃鹽酸,升溫至80℃回流6小時;取樣送LC-MS檢測顯示反應結束;
將產物分離提純:將反應液過濾,濾餅用25mL乙酸乙酯洗滌兩次,減壓濃縮,除去殘留的溶劑,得到雷迪帕韋關鍵中間體(具有式Ⅷ所示結構)1.87g,收率94%,純度97%;
使用質譜和核磁共振對產物結構進行檢測,可得譜圖數據為:MS=288[M/2+1]。1HNMR(400MHz CDCl3)δ10.88–10.62(m,1H),10.40–10.32(m,2H),8.37(s,1H),8.29–8.24(m,1H),8.12–7.93(m,5H),7.83–7.70(m,3H),5.32-5.23(m,1H),4.84(s,1H),4.20(s,2H),3.69-3.63(m,1H),3.19–3.11(m,2H),2.90–2.80(m,1H),2.29–2.21(m,1H),2.15–2.05(m,1H),2.04–1.92(m,1H),1.72–1.62(m,3H),0.88–0.82(m,2H),0.75-0.68(m,2H).
根據檢測結果可知,所得產物確實為具有式Ⅷ所示結構的化合物。
(6)向2L反應釜內加1000g DMF,87.8g的EDC.HCl,41.2g的HOBt,和80g MOC-L-valine,在25℃下攪拌30分鐘,降溫到0℃,加入100克具有式Ⅷ所示結構的化合物,在0℃下滴加100g的N-甲基嗎啉,30分鐘加完;加完后,升溫到25℃反應2小時,送樣檢測HPLC,VIII的含量在0.3%以內為反應終點,否則繼續反應到合格為止;
將產物分離提純:反應結束后,將反應液轉入5L反應瓶中,向反應液中加入2000g的乙酸乙酯和800g水,攪拌30分鐘后,靜置分層得有機相;水層用1440g的乙酸乙酯提取2次,將得到的有機相合并,用5%碳酸氫鈉溶液2000g洗滌有機相一次,1000g的水洗滌有機相一次,1000g的飽和氯化鈉溶液洗滌有機相一次;洗滌完成后向有機相中加入380g的元明粉和380g的硅膠,在25℃下攪拌1小時,過濾后濃縮濾液至無明顯液滴(240克左右);向濃縮物中慢慢滴加1120g的丙酮(1小時加完),降溫到10℃,攪拌24h,過濾后將產物在50℃下真空干燥至水分小于0.1%,得到雷迪帕韋粗品;粗品加入3倍重量的乙酸乙酯溶解后,過濾,在45℃下將濾液濃縮至干,加入11倍重量的丙酮溶解澄清后,攪拌10小時,慢慢析出固體,過濾得到產物雷迪帕韋,為白色粉末,收率為77.1%,純度為99.5%。
使用質譜和核磁共振對產物結構進行檢測,可得譜圖數據為:ESI-MS m/z:889[M+H]+;1H NMR(400MHz,DMSO)δ12.19(s,1H),11.85(s,1H),8.08(s, 1H),7.96(s,2H),7.92–7.75(m,4H),7.70(s,1H),7.64–7.48(m,2H),7.30(d,J=8.3Hz,1H),7.21(d,J=8.6Hz,1H),5.20(dd,J=7.7,5.1Hz,1H),4.67(d,J=5.9Hz,1H),4.55(s,1H),4.17(t,J=7.9Hz,1H),4.01(t,J=8.2Hz,1H),3.82(d,J=9.7Hz,1H),3.72(d,J=9.7Hz,1H),3.55(d,J=2.5Hz,6H),2.67(s,1H),2.41(d,J=9.3Hz,1H),2.22(m,1H),2.10–1.69(m,7H),1.51(m,2H),0.95(dd,J=15.5,6.5Hz,6H),0.90–0.75(m,7H),0.70(m,1H),0.66–0.46(m,3H).13C NMR(101MHz,CDCl3)δ170.50,170.00,156.93,155.37,149.42,143.86,143.14,141.84,138.30,137.98,137.74,137.49,136.25,135.95,134.64,133.97,133.26,131.18,127.95,125.77,123.35,121.86,121.30,120.96,119.31,116.77,113.60,111.74,109.43,60.82,58.08,57.86,56.24,54.97,54.43,51.52,51.49,30.99,30.14,29.84,27.10,21.16,19.32,19.09,18.45,18.19,10.77,9.59.
根據檢測結果可知,所得產物確實為雷迪帕韋。
對整個反應過程中N-氟代雙苯磺酰胺的利用率進行計算,可得N-氟代雙苯磺酰胺的利用率為59.4%。
實施例2
(1)在1L的四口瓶中加入20.0g具有式Ⅰ所示結構的化合物和400mL四氫呋喃,降溫至5℃,加入10.2g叔丁醇鉀,滴加13.2g 2-(三甲基硅烷基)乙氧甲基氯,滴加過程溫度控制在5℃,滴加結束后自然升溫至室溫,攪拌反應4小時;反應完成后取樣,TLC(PE:EA=1:1)檢測顯示反應結束;
按照實施例1中的方法對產物進行分離提純,得到具有式Ⅱ所示結構的化合物23.3g,收率78%,純度98%;
(2)在2.0L反應瓶中加入70g 2-溴-7-氯-9H-芴,190g N-氟代雙苯磺酰胺和700mL四氫呋喃,在氮氣保護下,反應液降溫至-60℃,緩慢滴加700mL1.0mol/L的雙(三甲基硅烷基)氨基鋰的四氫呋喃溶液,滴加時溫度控制在-60℃,滴加結束后,在-60℃下攪拌反應1.5小時;取樣送LC-MS檢測顯示反應結束;
按照實施例1中的方法對產物進行分離提純,得到得51.5g 2-溴-7-氯-9,9-二氟-9H-芴;收率65.2%,純度98.6%;
(3)在氮氣保護下,向500mL單口瓶中加入12.2g具有式Ⅱ所示結構的化合物,8.1g 2-溴-7-氯-9,9-二氟-9H-芴,16.7g碳酸銫,290mg Pd(pph3)4,1100mg SPhos和200mL N-甲基吡咯烷酮,加完后,反應體系升溫至130℃攪拌反應11小時;取樣TLC(PE:EA=3:1)檢測顯示反應結束;
按照實施例1中的方法將產物進行分離提純,得到具有式Ⅴ所示結構的化合物的粗品,粗產品可直接用于下一步反應。
(4)在500mL單口瓶中加入8.0g具有式Ⅴ所示結構的化合物,6.5g具有式Ⅵ所示結構的化合物,2.1g碳酸氫鈉,290mg Pd(dppf)Cl2,1050mgSPhos,200mL乙二醇二甲醚;反應體系用氮氣置換三次,升溫至110℃回流6小時;取樣送LC-MS檢測顯示反應結束;
按照實施例1中的方法將產物分離提純,得到具有式Ⅶ所示結構的化合物10.3g,收率89.5%,產物純度98%;
(5)在250mL單口瓶中加入5g具有式Ⅶ所示結構的化合物和100mL丙酮,滴加3.8g氫溴酸,升溫至70℃回流5小時;取樣送LC-MS檢測顯示反應結束;
按照實施例1中的方法將產物分離提純,得到雷迪帕韋關鍵中間體(具有式Ⅷ所示結構的氫溴酸鹽)4.86g,收率98%,純度98.5%;
(6)向2L反應釜內加1000g二氯甲烷,166g的HATU和165g MOC-L-valine,在20℃下攪拌30分鐘,降溫到0℃,加入200克具有式Ⅷ所示結構的化合物,在0℃下滴加200g的三乙胺,30分鐘加完;加完后,升溫到20℃反應3小時,送樣檢測HPLC,有式Ⅷ所示結構的化合物的含量在0.3%以內為反應終點,否則繼續反應到合格為止;
按照實施例1中的方法將產物分離提純,得到產物雷迪帕韋,為白色粉末,收率為80.1%,純度為99.4%。
對整個反應過程中N-氟代雙苯磺酰胺的利用率進行計算,可得N-氟代雙苯磺酰胺的利用率為70.0%。
實施例3
(1)在2L的四口瓶中加入30.0g具有式Ⅰ所示結構的化合物和600mL四氫呋喃,降溫至3℃,加入15g叔丁醇鈉,滴加19.8g 2-(三甲基硅烷基)乙氧甲基氯,滴加過程溫度控制在3℃,滴加結束后自然升溫至室溫,攪拌反應3小時;反應完成后取樣,TLC(PE:EA=1:1)檢測顯示反應結束;
按照實施例1中的方法對產物進行分離提純,得到具有式Ⅱ所示結構的化合物36.3g,收率81%,純度98%;
(2)在2.0L反應瓶中加入50g 2-溴-7-氯-9H-芴,136g N-氟代雙苯磺酰胺和500mL異丙醚,在氮氣保護下,反應液降溫至-65℃,緩慢滴加500mL1.0mol/L的雙(三甲基硅烷基)氨基鉀的異丙醚溶液,滴加時溫度控制在-65℃,滴加結束后,在-65℃下攪拌反應2.5小時;取樣送LC-MS檢測顯示反應結束;
按照實施例1中的方法對產物進行分離提純,得到得37.4g2-溴-7-氯-9,9-二氟-9H-芴;收率66.2%,純度98.9%;
(3)在1000mL單口瓶中加入45.4g具有式Ⅱ所示結構的化合物,30g2-溴-7-氯-9,9-二氟-9H-芴,26.3g碳酸鉀,1.1g Pd(OAc)2,4.0g SPhos和600mL N,N-二甲基乙酰胺,氮氣置換三次,升溫至120℃回流反應5小時,取樣送LC-MS檢測顯示反應結束;
緩慢降溫至20℃,向反應混合液中加入50.3g具有式Ⅵ所示結構的化合物和3.5g Pd(dppf)Cl2,氮氣置換三次,升溫至120℃回流7小時,取樣送LC-MS檢測顯示反應結束;
按照實施例1中的方法將產物分離提純,得到具有式Ⅶ所示結構的化合物53.3g,收率62%,產物純度98%;
(4)在500mL單口瓶中加入10g具有式Ⅶ所示結構的化合物和200mL丙酮,滴加7.6g氫溴酸,升溫至75℃回流6小時;取樣送LC-MS檢測顯示反應結束;
按照實施例1中的方法將產物分離提純,得到雷迪帕韋關鍵中間體(具有式Ⅷ所示結構的氫溴酸鹽)7.0g,收率98.5%,純度98.9%;
(5)向2L反應釜內加1000g二氯甲烷,200g的HATU和200g MOC-L-valine,在20℃下攪拌30分鐘,降溫到0℃,加入250克具有式Ⅷ所示結構的化合物,在0℃下滴加250g的三乙胺,30分鐘加完;加完后,升溫到20℃反應3小時,送樣檢測HPLC,具有式Ⅷ所示結構的化合物的含量在0.3%以內為反應終點,否則繼續反應到合格為止;
按照實施例1中的方法將產物分離提純,得到產物雷迪帕韋253.3g,為白色粉末,收率為82.1%,純度為99.5%。
對整個反應過程中N-氟代雙苯磺酰胺的利用率進行計算,可得N-氟代雙苯磺酰胺的利用率為50.0%。
由以上實施例可知,本發明提供的制備方法簡單、步驟少,關鍵化合物2-溴-7-氯-9,9-二氟-9H-芴(式Ⅳ)僅需經過碳-氫活化偶聯反應、Suzuki偶聯反應和去保護基反應,三步即可得到制備雷迪帕韋的關鍵中間體VIII,且可以通過“一鍋法”實現碳-氫活化反應和Suzuki偶聯反應,提高了氟化合物的利用率,降低了制備成本。
以上所述僅是本發明的優選實施方式,應當指出,對于本技術領域的普通技術人員來說,在不脫離本發明原理的前提下,還可以做出若干改進和潤飾,這些改進和潤飾也應視為本發明的保護范圍。
- 還沒有人留言評論。精彩留言會獲得點贊!