使用切口酶的解旋酶依賴性等溫擴增的制作方法
本申請為國際申請日2011年8月16日、國際申請號pct/ep2011/064114于2013年2月16日進入中國國家階段、申請號201180039588.6、發明名稱“使用切口酶的解旋酶依賴性等溫擴增”的分案申請。本發明屬于生物學和化學領域,特別是屬于分子生物學領域。更具體地,本發明涉及用于擴增核酸的方法和試劑盒。
背景技術:
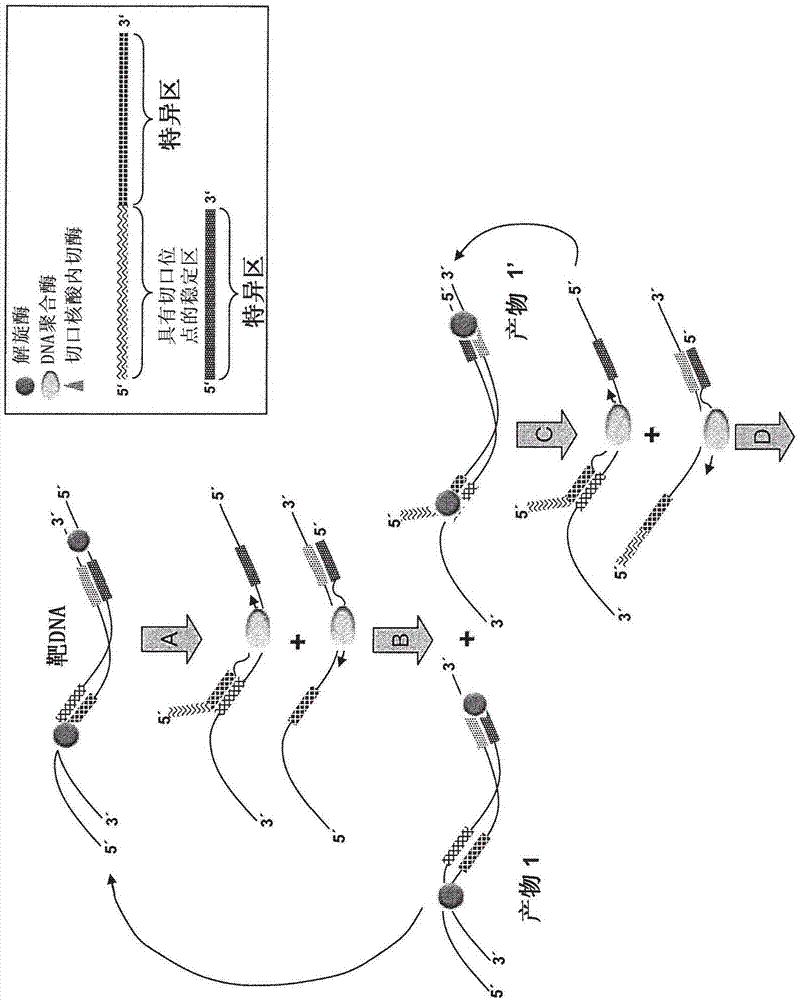
:核酸擴增被廣泛用在研究、診斷學、法醫學、醫學、食物科學和農業中。“現場即時測試(pointofcaretesting)”(poct)是指在患者醫療點處或附近進行的診斷性測試,即去集中化的檢查,例如可以在私人醫療實踐、小醫院、藥店中或者甚至在事故或其它醫療緊急事件的現場或位點處或者在救護車內執行所述檢查。現場即時診斷需要快速且簡單的測試。因此,在基于核酸的檢測方法的情況下,與已建立的但較慢的基于pcr的方法相比,快速等溫擴增技術最近已變得越來越重要。pcr需要熱循環,以便將雙鏈核酸例如dna的兩條鏈分離開。相反,例如等溫擴增方法不需要熱循環儀。最杰出的等溫擴增方法之一是所謂的解旋酶依賴性擴增(hda)(例如被描述在vincent等.(2004),emboreports5(8):795-800;jeong等.(2009),cell.mol.lifesci66:3325-3336;wo-a22004/027025;wo-a22006/074334中,所有均通過參考結合于此)。hda是基于解旋酶將雙鏈核酸特別是dna解旋的能力,而不需要加熱或甚至熱循環。在hda中,使用dna聚合酶和合適的寡核苷酸引物來復制分離的dna鏈。因此,hda模擬天然的復制叉機制。hda需要存在atp、二價鎂離子和dntp。某些基于hda的方法還采用單鏈結合蛋白(ssb)來包被置換的dna鏈。事實上,取決于所使用的酶,可以在熱不穩定或熱穩定的反應中執行hda方法。典型地,在25℃和50℃之間、優選在37℃和42℃之間的溫度下執行熱不穩定的hda反應。相反,在超過50℃、典型地在60℃和70℃之間的溫度下執行熱穩定的hda或嗜熱的hda(thda)反應。hda反應選擇性地擴增由兩個引物限定的靶序列。hda使用解旋酶而不是像pcr中那樣使用熱來將雙鏈核酸的兩條鏈分離開。因此,可以在單個溫度下執行hda,而不需要熱循環。常規的標準hda反應的步驟示于圖1中:在第一個步驟中,通過解旋酶將雙鏈核酸解旋,產生(部分)單鏈的序列。然后,引物與單鏈區結合。在步驟2中,聚合酶合成互補鏈。最后,解旋酶和聚合酶共同起作用,導致模板的擴增(步驟3)。其它等溫擴增包括鏈置換擴增(sda),鏈置換擴增是基于使用限制性酶在dna的未修飾鏈上造成切口并通過核酸外切酶缺陷的dna聚合酶的作用來延伸切口處的3’末端以置換下游的dna鏈。另一種等溫擴增是滾環擴增(rca),其中線性ssdna被退火到環狀ssdna模板上,所述環狀ssdna模板是通過使用dna連接酶連接模板dna的兩個末端而生成的。接著,使用dna聚合酶延伸退火的引物,并生成互補模板序列的串聯連接的拷貝。rca和sda兩者都需要初始的熱變性步驟。其它等溫擴增包括例如切口酶擴增反應(near)和相關的指數擴增反應(expar),兩者都采用切口酶和聚合酶來擴增短的雙鏈模板序列(例如被描述在vanness等.(2003),proc.natl.acad.sci.100(8):4504-4509;us-a12003/0082590;wo-a22004/022701;wo-a22009/012246;wo-a22004/067726中,所有均通過參考結合于此)。然而,所有上述等溫擴增方法均需要復雜的反應方案,是相對慢的和/或僅限于相對短的模板序列。技術實現要素:本發明提供了用于擴增核酸、優選雙鏈核酸的改進的方法,所述方法克服了現有技術的這些限制。本發明的方法基于改良的hda方法。所提供的方法比常規的thda快,同時具有可靠的特異性。與類似于near和expar的方法相比,本發明的方法可以擴增更長的模板核酸。本發明涉及在切口核酸內切酶存在下執行的hda擴增方法。因此,在本發明的情形下,在解旋酶、合適的聚合酶、和切口核酸內切酶存在下擴增模板核酸。具體地,本發明涉及用于擴增模板核酸、特別是dna的方法,其中所述方法包括在切口核酸內切酶存在下使用解旋酶依賴性擴增(hda)反應、特別是嗜熱的hda(thda)擴增所述模板核酸,并且其中所述模板核酸包含由所述切口核酸內切酶識別的序列,或者在hda反應期間由所述切口核酸內切酶識別的序列被引入到模板核酸中。可以使用包含由切口核酸內切酶識別的序列的寡核苷酸引物將由切口核酸內切酶識別的序列引入到模板核酸中。這樣的引物可以包含不與模板核酸雜交但是包含由切口核酸內切酶識別的序列或其部分的5’標簽序列。本發明還涉及用于擴增核酸的試劑盒,所述試劑盒包含-切口核酸內切酶,-解旋酶,和-dna聚合酶。本發明的方法和試劑盒可用于擴增核酸,也可用于檢測核酸和/或對核酸進行定量。附圖說明圖1示出了現有技術的且可商購的例如來自于biohelix/newenglandbiolabs的標準thda反應、即在切口核酸內切酶不存在下的thda反應的方案。圖2示出了本發明的使用帶標簽引物的切口thda的具體實施方案的反應方案。圖3示出了本發明的使用不帶標簽引物的切口thda的具體實施方案的反應方案。圖4示出了在管掃描器中來自于實施例1的不同的實時(切口)thda反應的結果(擴增曲線)。圖中所示的是隨時間推移的原始熒光強度。圖5示出了實施例1的擴增產物的熔融曲線。a)標準thda,b)無nb.bsmi的切口thda,c)有nb.bsmi的切口thda。圖6示出了具有擴增和電泳后的實施例1的各個反應混合物的聚丙烯酰胺凝膠。a)染色前的凝膠,b)用etbr染色后的凝膠。c)凝膠上各個條帶b1至b5的內含物還被圖示在圖6b1至6b5中。圖7示出了實施例2的靶dna連同所使用的引物的序列。圖8示出了反應速率對切口核酸內切酶的濃度的依賴性。圖中示出的是在可以檢測到顯著高于背景信號的擴增子信號之前的以分鐘(min)為單位的時間。在存在較低濃度的切口核酸內切酶時,比無切口核酸內切酶的反應早約5min檢測到信號,表示反應更快。圖9示出了標準thda即無任何切口核酸內切酶的thda的擴增曲線(隨時間推移的熒光)。在cdna模板存在下測得三條曲線,在cdna模板不存在下測得兩條曲線。圖10示出了在0.1u切口核酸內切酶nt.bstnbi存在下thda的擴增曲線。在cdna模板存在下測得三條曲線,在cdna模板不存在下測得兩條曲線。圖11示出了在無切口核酸內切酶nt.bstnbi的標準thda之后的熔融曲線分析。對圖9中示出的反應進行熔融曲線分析。在cdna模板存在下測得三條曲線,在cdna模板不存在下測得兩條曲線。圖12示出了在0.1u切口核酸內切酶nt.bstnbi存在下標準thda之后的熔融曲線分析。對圖10中示出的反應進行熔融曲線分析。在cdna模板存在下測得三條曲線,在cdna模板不存在下測得兩條曲線。圖13示出了反應速率對切口核酸內切酶的存在的依賴性。圖中示出的是在可以檢測到顯著高于背景信號的擴增子信號之前的以分鐘(min)為單位的時間。在切口核酸內切酶存在下,比無切口核酸內切酶的反應早約6min檢測到信號,表示反應更快。圖14示出了實施例1的擴增子的一條鏈的序列,實施例1的擴增子是指淋病奈瑟氏菌(neisseriagonorrhoeae)基因組的孔蛋白基因的一部分。指出了引物和探針的雜交區。(a):標準thda;(b):本發明的方法的使用帶標簽引物的切口thda,帶下劃線的是nb.bsmi的識別序列,由帶標簽引物引入的序列是斜體的。具體實施方式本發明提供了用于擴增并任選地檢測核酸、特別是dna的方法和試劑盒。本發明的方法基于解旋酶依賴性擴增(hda)反應。然而,不同于現有技術的hda反應,在本發明的擴增反應中存在切口核酸內切酶。因此,本發明的試劑盒也包含切口核酸內切酶。本發明的方法在本文中可以被稱為“切口hda”。具體地,本發明的方法和試劑盒可用于擴增和檢測短dna序列,優選小于400bp的序列,更優選小于200bp的序列,甚至更優選150bp以下的序列,最優選在70bp和120bp之間的序列。典型地,用本發明的方法和試劑盒,擴增或檢測雙鏈核酸。然而,也可以擴增和檢測單鏈核酸,只要在實際的基于hda的擴增開始之前用聚合酶將它們轉錄成核酸雙鏈即雙鏈模板即可。具體地,本發明涉及用于擴增模板核酸的方法,其中所述方法包括在切口核酸內切酶存在下使用解旋酶依賴性擴增(hda)反應擴增所述模板核酸,并且其中所述模板核酸包含由所述切口核酸內切酶識別的序列,或者在hda反應期間由所述切口核酸內切酶識別的序列被引入到模板核酸中。可以采用多種模板核酸、優選采用雙鏈核酸來使用所述方法和試劑盒。典型地,用本發明的試劑盒和方法擴增和/或檢測dna。因此,在本發明的情形下,模板核酸優選為雙鏈核酸,更優選為dna。例如,待擴增或檢測的序列可以是線性dna或環狀dna。dna例如可以選自基因組dna,例如微生物基因組dna、病毒dna、質粒dna和cdna。模板可以反轉錄自rna,特別是mrna。在本發明的情形下,“dsdna”和“dna”雙鏈是指雙鏈dna,“ssdna”是指單鏈dna,“dsrna”是指雙鏈rna,“ssrna”是指單鏈rna。還可以用本發明的試劑盒和方法擴增其它核酸,例如dsrna或dna/rna雜交體。可以在hda之前執行其它的酶促步驟例如反轉錄或體外轉錄,以形成可用的核酸。本發明要求待擴增的模板核酸中存在由切口核酸內切酶識別的序列。由于不是每個待擴增的靶序列都包含這樣的切口核酸內切酶識別序列,因此,可以通過合適的引物來引入這樣的序列。因此,在本發明的方法的優選實施方案中,使用包含由切口核酸內切酶識別的序列的寡核苷酸引物將由切口核酸內切酶識別的序列引入到模板核酸中。在某些實施方案中,包含由切口核酸內切酶識別的序列的引物包含不與模板核酸雜交的5’標簽序列,并且由切口核酸內切酶識別的序列處于所述標簽序列中。這樣的引物在本文中被稱為“帶標簽引物”,圖2中示意性示出了對應的實施方案。還可以采用誘變引物,即,與原始模板序列不是100%互補的引物,由此引入合適的切口核酸內切酶的識別序列。本發明的hda反應優選為嗜熱的hda(thda),即,所使用的酶在所述方法的溫度下是熱穩定的。熱穩定的酶典型地為源自于熱穩定的生物的酶。因此,優選地,在超過60℃、更優選在60℃和70℃之間、甚至更優選在60℃和65℃之間、最優選在約64℃至65℃的溫度下執行本發明的方法。例如可以使用水浴、加熱模塊、溫箱或熱循環儀來維持溫度。本發明的方法通常為等溫方法,這意味著,該方法是在單個溫度下執行的,而不像例如在pcr中那樣需要溫度循環。然而,在本發明的某些實施方案中,在恒定的實際反應溫度下執行反應之前,可以包括在典型地超過90℃、優選約95℃的高溫下的熱變性步驟。于是,與在單個溫度下執行的“一步法”方案不同,這被稱為“兩步法”反應。在某些情況下,兩步法方案可以產生更高的靈敏度。因此,在本發明的情形下,兩步法方案是優選的。事實上,在本發明的切口hda方法中,在解旋酶、聚合酶、切口酶以及合適的正向和反向寡核苷酸引物存在下溫育模板核酸。另外,需要存在用于擴增反應的其它試劑,例如dntp、atp或datp以及鎂離子。反應混合物優選地還包含緩沖劑和鹽例如nacl和/kcl。各個酶和試劑可以在開始擴增之前被一起加入或者可以被順次加入。在某些具體實施方案中,在其它組分之后加入切口核酸內切酶。在兩步法方案的優選實施方案中,在初始的熱變性步驟之后加入酶。術語“引物”是指能夠與靶核酸上的單鏈區結合以促進靶核酸的聚合酶依賴性復制的單鏈核酸。在本發明的情形下,向模板核酸添加一個或多個引物。使用一個引物引起線性擴增,而使用兩個或甚至更多個引物引起指數擴增。術語“解旋酶”在本文中是指能夠將雙鏈核酸酶促解旋的任何酶。例如,解旋酶是可見于所有生物以及所有涉及核酸的過程例如復制、重組、修復、轉錄、翻譯和rna剪接的酶。(kornberg和baker,《dna復制》(dnareplication),w.h.freemanandcompany(第2版(1992)),特別是第11章)。任何沿著dna或rna以5’至3’方向或以相反的3’至5’方向易位的解旋酶均可用于本發明的實施方案。這包括從原核生物、病毒、古生菌和真核生物獲得的解旋酶或天然存在的酶的重組形式,以及具有指定的活性的類似物或衍生物。由kornberg和baker在他們的書《dna復制》,w.h.freemanandcompany(第2版(1992))的第11章中描述的天然存在的dna解旋酶的實例包括大腸桿菌(e.coli)解旋酶i、ii、iii和iv,rep,dnab,pria,pcra,t4gp41解旋酶,t4dda解旋酶,t7gp4解旋酶,sv40大t抗原,酵母rad。可用于hda的其它解旋酶包括recq解旋酶(harmon和kowalczykowski,j.biol,chem.276:232-243(2001))、來自于騰沖嗜熱菌(t.tengcongensis)和極端嗜熱菌(t.thermophilus)的熱穩定的uvrd解旋酶(collins和mccarthy,extremophiles.7:35-41.(2003))、來自于水生嗜熱菌(t.aquaticus)的熱穩定的dnab解旋酶(kaplan和steitz,j.biol.chem.274:6889-6897(1999))、以及來自于古生菌和真核生物的mcm解旋酶(grainge等,nucleicacidsres.31:4888-4898(2003))。通常以5’至3’方向復制的解旋酶的實例是t7gp4解旋酶、dnab解旋酶和rho解旋酶,而以3’-5’方向復制的解旋酶的實例包括uvrd解旋酶、pcra、rep、hcv的ns3rna解旋酶。解旋酶可能需要輔因子,即,解旋酶的解旋活性所需要的小分子劑。解旋酶的輔因子包括三磷酸核苷(ntp)和脫氧核苷三磷酸(dntp濃度)和鎂(或其它二價陽離子)。例如,濃度在0.1-100mm范圍內、優選在1-10mm范圍內(例如3mm)的atp(三磷酸腺苷)可以用作uvrd解旋酶的輔因子。類似地,在1-10mm范圍內(例如3mm)的dttp(脫氧胸苷三磷酸)可以用作t7gp4b解旋酶的輔因子。在本發明的某些實施方案中,特別是對于非嗜熱的hda來說,反應中可以存在其它蛋白例如拓撲異構酶或輔助蛋白例如單鏈dna結合蛋白(ssb),以便促進解旋酶活性。在單鏈結合蛋白(ssb)存在下,解旋酶顯示出改進的活性。在這些情況下,ssb的選擇通常不限于特定的蛋白。單鏈結合蛋白的實例是t4基因32蛋白、大腸桿菌ssb、t7gp2.5ssb、噬菌體phi29ssb(kornberg和baker,同上(1992))和前述蛋白的截短形式。在使用熱穩定的解旋酶的情況下,一種或多種ssb或其它輔助蛋白的存在是任選的。在ep-b11539979中,特別是在[0039]至[0066]章節中更詳細地描述了用于hda和thda中的解旋酶和包含解旋酶的制劑。優選地,在超過50℃、優選在50℃和70℃之間、更優選在60℃和65℃之間的溫度下執行本發明的切口hda擴增反應。在本發明的情形下,解旋酶可以來自于不同的解旋酶家族。解旋酶優選選自超家族i解旋酶、超家族ii解旋酶、超家族iii解旋酶、來自于dnab樣超家族的解旋酶或者來自于rho樣超家族的解旋酶。解旋酶可以源自于嗜中溫或耐熱的生物。解旋酶可以是遺傳或化學修飾的。超家族i解旋酶例如包括dda、pcra、f質粒trai蛋白解旋酶和uvrd。超家族ii解旋酶例如包括recq和ns3解旋酶。超家族iii解旋酶例如包括aavrep解旋酶。來自于dnab樣超家族的解旋酶例如包括t7噬菌體解旋酶。在本發明的方法和試劑盒的情形下,優選的是,解旋酶是熱穩定的解旋酶。熱穩定的tte-uvrd解旋酶是優選的。拓撲異構酶可用于長hda反應,以提高hda擴增長的靶擴增子的能力。當解旋酶將非常長的線性dna雙鏈分離開時,拓撲異構酶的旋轉(松弛)功能消除纏繞并防止過旋(kornberg和baker,同上(1992))。例如,大腸桿菌拓撲異構酶i(fermentas,vilnius,lithuania)可通過在一條dna鏈中引入切口而用于松弛負超螺旋dna。相反,大腸桿菌dna促旋酶(拓撲異構酶ii)在dna中引入瞬時的雙鏈斷裂,從而允許dna鏈相互穿過(kornberg和baker,同上(1992))。原則上,可以選擇在所需的反應溫度下有活性、即能夠延長游離3’-oh末端的所有聚合酶。具有附加功能例如校對功能、核酸內切酶活性、鏈置換活性和/或反轉錄酶活性的聚合酶也可用于本發明的情形。基于持續性和鏈置換活性,選擇用于hda的聚合酶。在熔融并與引物雜交之后,使核酸經受聚合步驟。如果待擴增的核酸是dna,則選擇dna聚合酶。當初始靶標是rna時,則首先使用反轉錄酶將rna靶標復制成cdna分子,然后通過所選擇的dna聚合酶在hda中進一步擴增cdna。dna聚合酶作用于靶核酸,以在4種dntp存在下延伸與核酸模板雜交的引物,從而形成與核酸模板上的核苷酸序列互補的引物延伸產物。dna聚合酶選自缺乏5’至3’核酸外切酶活性的聚合酶,并且所述聚合酶還可以任選地缺乏3’-5’核酸外切酶活性。合適的dna聚合酶的實例包括大腸桿菌dna聚合酶i的核酸外切酶缺陷的klenow片段(newenglandbiolabs,inc.(beverly,ma))、核酸外切酶缺陷的t7dna聚合酶(測序酶;usb(cleveland,oh))、大腸桿菌dna聚合酶i的klenow片段(newenglandbiolabs,inc.(beverly,ma))、bstdna聚合酶的大片段(newenglandbiolabs,inc.(beverly,ma))、klentaqdna聚合酶(abpeptides(stlouis,mo))、t5dna聚合酶(美國專利no.5,716,819)、和poliiidna聚合酶(美國專利no.6,555,349)。在本發明的情形下,優選的是具有鏈置換活性的dna聚合酶,例如大腸桿菌dna聚合酶i的核酸外切酶缺陷的klenow片段、bstdna聚合酶的大片段和測序酶。t7聚合酶是高保真的聚合酶,出錯率為3.5x105,顯著低于tag聚合酶(keohavong和thilly,proc.natl.acad.sci.usa86,9253-9257(1989))。然而,t7聚合酶不是熱穩定的,因此,對于在需要熱循環的擴增系統中的使用而言,t7聚合酶不是最佳的。在可以等溫執行的hda中,t7測序酶是用于擴增dna的優選聚合酶之一。在本發明的方法和試劑盒的情形下,聚合酶優選為熱穩定的聚合酶。優選地,聚合酶選自bstdna聚合酶、pyrophage聚合酶、displaceace聚合酶和vent(exo-)聚合酶。沒有顯著的核酸外切酶活性的聚合酶是優選的。一般來說,適合用于hda和切口hda的引物是短的合成寡核苷酸,例如具有大于10個核苷酸且小于50個核苷酸的長度。寡核苷酸引物的設計涉及多種參數,例如基于鏈的比對得分(string-basedalignmentscore)、熔融溫度、引物長度和gc含量(kampke等,bioinformatics17:214-225(2003))。在設計引物時,重要的因素之一是選擇的序列在待擴增的核酸分子特有的靶片段內。另一個重要因素是確定用于hda反應的引物的熔融溫度。引物的熔融溫度由寡核苷酸的長度和gc含量決定。優選地,引物的熔融溫度在會發生雜交和擴增的溫度范圍內。例如,如果當使用大腸桿菌uvrd解旋酶制劑時,雜交和擴增的溫度被設定在37℃,則被設計用于該反應的引物對的熔融溫度應在約30℃至50℃范圍內。如果雜交和擴增的溫度是60℃,則被設計用于該反應的引物對的熔融溫度應在55℃和70℃之間、優選在55℃和65℃之間的范圍內。熔融溫度顯著高于反應溫度的引物可能引起非特異性副反應,因此是不太優選的。為了選擇用于hda反應的最佳引物,可以在平行分析中測試一組具有各種不同的熔融溫度的引物。kampke等,bioinformatics17:214-225(2003)描述了關于引物設計的更多信息。各引物與靶核酸的每一末端雜交,并可以由聚合酶使用靶核苷酸序列作為模板以3’至5’方向延伸。雜交條件是如《分子克隆和實驗指南》(“molecularcloningandlaboratorymanual”)第二版,sambrook、rich和maniatis出版,coldspringharbor(2003)中描述的標準條件。為了實現特異性擴增,同源或完全匹配的引物是優選的。然而,引物可以在5’末端包含不與靶核苷酸序列互補的序列。或者,整個引物中都可以含有不與靶核酸精確互補的核苷酸或序列。引物可以表示類似的引物,或者可以是用于hda的非特異性引物或通用引物,只要可以在預定溫度下通過引物-模板結合實現特異性雜交即可。引物可以包含脫氧核糖核苷酸堿基a、t、g或c中的任一種和/或一個或多個核糖核苷酸堿基a、c、u、g和/或一個或多個修飾的核苷酸(脫氧核糖核苷酸或核糖核苷酸),其中修飾不會阻止引物與核酸的雜交或者引物的延長或者雙鏈分子的變性。可以用化學基團例如硫代磷酸酯或甲基膦酸酯或者用非核苷酸接頭修飾引物,以提高它們的性能或促進擴增產物的表征。為了檢測擴增產物,可以使引物經受修飾,例如熒光或化學發光標記和生物素化。其它標記方法包括放射性同位素、發色團和配體例如生物素或半抗原,所述配體盡管不能被直接檢測到,但可以通過與其特異性結合伙伴例如分別為抗生物素蛋白和抗體的標記形式反應而被容易地檢測到。可以通過本領域已知的方法制備如本文所述的引物。(參見,例如美國專利no.6,214,587)。在某些實施方案中,在本發明的方法中使用一對的兩個序列特異性引物來實現靶序列的指數擴增,其中一個引物與靶序列的5’邊界雜交,另一個引物與靶序列的3’邊界雜交。該方法可以容易地與lee等(j.mol.biol.316:19-34(2002))區分開。在單個hda反應中可以利用多對引物以擴增多個靶標,同時在多重反應中使用不同的檢測標簽。多重反應常用于snp分析以及病原體檢測(jessing等,j.clin.microbiol.41:4095-4100(2003))。例如可以使用primerquest程序(http://www.idtdna.com/scitools/applications/primerquest/advanced.aspx)或primer3程序(http://frodo.wi.mit.edu/cgi-bin/primer3/primer3_www.cgi)來設計thda引物。在本文中使用時,“切口”是指在相對于由執行切口的酶識別的核苷酸序列而言的特定位置處只對完全雙鏈的核酸分子或部分雙鏈的核酸分子的雙鏈部分的一條鏈進行的切割。其中核酸被切口的特定位置被稱為“切口位點”。“切口核酸內切酶”是識別完全或部分雙鏈的核酸分子的特定核苷酸序列并在相對于識別序列而言的特定位置處只切割核酸分子的一條鏈的酶。在本文中使用時,“切口核酸內切酶”是指識別完全或部分雙鏈的核酸分子的核苷酸序列并在相對于識別序列而言的特定位置處只切割核酸分子的一條鏈的核酸內切酶。限制性核酸內切酶典型地識別只由天然核苷酸構成的核苷酸序列,并只切割含有核苷酸序列的完全或部分雙鏈的核酸分子的一條鏈。在本發明的方法和試劑盒的情形下,切口核酸內切酶優選為熱穩定的切口核酸內切酶。切口核酸內切酶是可商購的,例如購自newenglandbiolabs(neb)。優選地,切口核酸內切酶選自nb.bsmi、nt.bstnbi、nt.cvipii、nb.bbvci、nb.bsrdi、nb.btsi、nt.bsmai、nt.bbvci、nt.bspqi和nt.alwi。nb.bsmi和nt.bstnbi是特別優選的切口核酸內切酶。例如通過遺傳工程改造標準的限制性核酸內切酶而獲得的切口核酸內切酶也可用于本發明的情形。nb.bsmi是只切割雙鏈dna底物上的一條dna鏈的切口核酸內切酶;它識別序列5’-gaatgc-3’并在互補鏈上如斜線所指示的進行切割:5’-g/cattc-3’。nt.bstnbi是位點特異性核酸內切酶,其只切割雙鏈dna底物上的一條dna鏈;它在其識別序列5’-gagtc-3’的3’側之外4個堿基處催化單鏈斷裂。根據本發明所使用的反應溫度,選擇在該溫度下有活性的合適的切口核酸內切酶。在“包含標簽序列的引物”(“帶標簽引物”)的情況下,“由切口核酸內切酶識別的序列”(“切口核酸內切酶識別序列”)可以完全或部分處于所述標簽序列中。切割位點也可以處于所述標簽序列中,或者它可以在上游或下游的一個或幾個堿基對(bp)處。切割位點(“切口位點”)可以在與所指示的識別序列相同的鏈上,或者在另一條鏈上。在一個實施方案中,本發明的方法可以被描述為包括以下步驟:(i)提供雙鏈模板核酸,(ii)使模板核酸與用于使雙鏈模板核酸解旋的解旋酶接觸,以形成至少部分單鏈的模板核酸,(iii)加入用于與步驟(ii)的單鏈模板雜交的寡核苷酸引物,其中至少一個寡核苷酸引物包含由切口核酸內切酶識別的序列,(iv)用聚合酶合成與單鏈模板核酸互補的寡核苷酸引物的延伸產物,以形成雙鏈模板核酸,(v)使在步驟(iv)中合成的雙鏈模板核酸與用于在模板核酸中引入切口的切口核酸內切酶接觸,(vi)使步驟(v)的模板核酸與使雙鏈模板核酸解旋的解旋酶接觸,以形成至少部分單鏈的模板核酸,(vii)加入用于與步驟(vi)的單鏈模板雜交的寡核苷酸引物,(viii)用聚合酶合成與單鏈模板核酸互補的寡核苷酸引物的延伸產物,以形成雙鏈模板核酸,(ix)根據需要重復步驟(v)至(viii),以擴增模板核酸。在擴增單鏈模板核酸的情況下,必須通過例如反轉錄或體外轉錄將單鏈模板核酸轉錄成雙鏈核酸。在本發明的方法的具體實施方案中,寡核苷酸引物的由切口核酸內切酶識別的序列處于不與模板核酸雜交的5’標簽序列中,并且所述方法在步驟(iv)和(v)之間還包括以下步驟:(a)使步驟(iv)的模板核酸與用于使雙鏈模板核酸解旋的解旋酶接觸,以形成至少部分單鏈的模板核酸,(b)加入用于與步驟(a)的單鏈模板雜交的寡核苷酸引物,(c)用聚合酶合成與單鏈模板核酸互補的寡核苷酸引物的延伸產物,以形成雙鏈模板核酸。在圖2中示意性地示出了在dna上使用帶標簽引物的切口thda反應的過程:步驟a:通過解旋酶將dsdna(“靶dna”,“模板dna”)解旋,并且引物對與單鏈dna結合。一個引物具有不與模板dna雜交的5’標簽序列。dna聚合酶開始合成從引物開始的互補鏈。步驟b:形成兩條雙鏈產物dna:基于不帶標簽引物的產物1對應于模板dna并再次進入步驟a中的反應;產物1’包含標簽序列并被解旋酶解旋。步驟c:通過dna聚合酶合成產物1’的互補鏈。步驟d:一條鏈產生產物1’并再次進入步驟c中的反應,另一條鏈產生產物2,產物2包含具有功能性切口位點和切口酶識別序列的標簽序列。步驟e:產物2的一條鏈在切口位點處被切割,并被解旋酶從另一末端開始解旋。dna聚合酶延長切割位點處的3’末端并置換鏈(“鏈置換擴增”(sda))。引物結合在另一條鏈上并被dna聚合酶延長。步驟f:引物延長產生了兩種產物,產物3包含標簽序列,產物3’不包含標簽序列。產物3在切口位點處被再次切割,并合成了互補鏈。產物3’被解旋酶解旋。步驟g1:產物3的擴增產生了再次進入步驟g1的產物3和進入步驟g2的產物3’。步驟g2:合成兩條ssdna鏈的互補鏈,產生了再次進入步驟e和g2中的反應的產物2和3’。總反應導致產物3的積累。可以用雜交探針例如與擴增子的反向鏈互補的雜交探針檢測該產物。在圖3中示意性地示出了在dna上使用不帶標簽引物的切口thda反應的過程。步驟a:向dsdna添加包含至少一個引物、dna聚合酶、解旋酶和切口核酸內切酶的反應混合物。引物包含切口核酸內切酶的識別序列。dsdna被解旋酶解旋,引物與單鏈雜交并被延長。在一條鏈中引入了切口核酸內切酶識別位點。步驟b:所產生的dsdna被切口核酸內切酶切割。步驟c:解旋酶將dsdna解旋,舊的引物或新結合的引物在3’末端處被dna聚合酶延長。在下文中,總結了dna的一步法和兩步法切口thda反應的典型的但僅僅是示例性的并且是非限制性的方案:兩步法thda方案1.向包含dntp、datp或atp,鎂離子(例如硫酸鎂mgso4),緩沖劑例如trishcl,以及鹽例如nacl和/或kcl的合適的水性反應緩沖液中加入模板dna以及正向和反向引物。例如微量離心管類型可以被用作反應容器。2.將反應混合物加熱至變性溫度(例如95℃,持續例如2min),任選地在4℃下冷卻或置于冰上,然后在實際反應溫度下持續短時間(例如64℃或65℃,持續例如1至3min)。然后,任選地將反應混合物置于冰上或4℃。3.向反應混合物中加入酶,即解旋酶、dna聚合酶和切口酶。例如可以通過短暫的渦旋或通過移液管吹打輕輕地混合反應混合物。可以將酶預先混合在合適的緩沖液中。4.在合適的溫度下例如在64℃或65℃下將反應混合物溫育合適的時間,例如60至90min,優選為60至75min。一步法thda方案對于一步法方案而言,略過了兩步法方案的步驟2。在步驟1之后,直接進行到步驟3。用于本發明的方法的典型緩沖條件如下:-tris:5至100mm,優選為10mm-ph7.5至9.5,優選為ph8.8-kcl:0至50mm,優選為5mm-mgso4:1至6mm,優選為3.5mm-nacl:0至100mm,優選為40mm-dntp:0.1至1mm,優選為0.4mm-(d)atp(即datp和/或atp):1至7mm,優選為3mm-引物(正向):0.02至1.5μμ,優選為0.1μμ-引物(反向):0.02至1.5μμ,優選為0.1μμ-切口核酸內切酶:0.01-10u/反應,優選為0.5至3u/反應(25μl/反應)-聚合酶:0.3至3u/μl,優選為0.6至2u/μl-解旋酶:1至100ng/μl,優選為3至20ng/μl任選地可以加入甜菜堿,優選其濃度為100至2000mm,優選為700至1200mm。然而,這些只是示例性的條件,可以根據其它參數例如所使用的具體的酶、模板和/或引物序列或者溫度對所述條件進行調整。在具體實施方案中,本發明的方法還包括檢測擴增產物的步驟。例如可以使用凝膠電泳、嵌入染料或特異性寡核苷酸探針來檢測擴增產物。可通過多種方法來檢測擴增的核酸產物,所述方法包括溴化乙錠染色以及用選自以下的標簽檢測擴增的序列:放射性標簽、熒光標簽和酶。例如可以使用熒光標記的luxtm引物(invitrogencorporation,carlsbad,ca)實時檢測hda擴增產物,所述引物是在3’末端附近設計有熒光團的發夾結構的寡核苷酸。該構型固有地賦予了熒光猝滅能力,而無需單獨的淬滅基團。當引物變成被摻入到雙鏈擴增產物中時,熒光團被去淬滅化,導致熒光信號顯著增加。寡核苷酸探針優選包含熒光標簽,即典型地為共價連接的熒光染料。具體地,熒光標記的探針可以標記有選自以下的染料:fam、vic、ned、熒光素、fitc、ird-700/800、cy3、cy5、cy3.5、cy5.5、hex、tet、tamra、joe、rox、bodipytmr、俄勒岡綠、羅丹明綠、羅丹明紅、德克薩斯紅、亞基馬黃、alexafluor和pet。合適的雜交探針包括lightcycler探針(roche)、taqman探針(roche)、分子信標、scorpion引物、sunrise引物、lux引物和amplifluor引物。在本發明的情形下,taqmn探針是優選的。本發明還涉及用于擴增核酸的試劑盒,所述試劑盒包含-切口核酸內切酶,-解旋酶,和-dna聚合酶。對于本發明的方法而言,試劑盒的解旋酶優選選自:超家族i解旋酶,例如dda、pcra、f質粒trai蛋白解旋酶和uvrd;超家族ii解旋酶,例如recq和ns3解旋酶;超家族iii解旋酶,例如aavrep解旋酶;來自于dnab樣超家族的解旋酶,例如t7噬菌體解旋酶;以及來自于rho樣超家族的解旋酶。優選的是,解旋酶是熱穩定的解旋酶。熱穩定的tte-uvrd解旋酶是特別優選的。試劑盒的聚合酶優選自bstdna聚合酶、pyrophage聚合酶、displaceace聚合酶和vent(exo-)聚合酶。試劑盒的切口核酸內切酶優選自nb.bsmi、nt.bstnbi、nt.cvipii、nb.bbvci、nb.bsrdi、nb.btsi、nt.bsmai、nt.bbvci、nt.bspqi和nt.alwi。本發明的試劑盒還可以包含-緩沖劑,例如tris,和/或-dntp,和/或-(d)atp,和/或-鎂鹽,例如磷酸鎂,和/或-nacl,和/或-kcl。本發明的試劑盒還可以包含拓撲異構酶和/或ssb。試劑盒的各個組分可以在一個或多個容器中。兩個或更多個組分可以例如共同在一個容器例如反應管或小瓶中。優選地,緩沖劑、鹽、dntp以及如果存在的話還有(d)atp在預先混合的溶液中。所述試劑盒還可以包含手冊。本發明還涉及本發明的方法和試劑盒在擴增和/或檢測核酸中的應用。所述方法和試劑盒例如可以用于檢測微生物、病毒、病原體、人類的基因組dna和cdna等。對其中的核酸進行檢測的樣品例如可以是臨床樣品例如體液,例如血液、血漿、血清和尿液;或者可以是來自于環境、植物、動物、牲畜、食品的樣品或來自于犯罪現場的樣品。所述方法和試劑盒例如可以用于專用的實驗室、護理點或現場。核酸可以在擴增之前從樣品中被純化和/或分離,或者它們可以在樣品中被直接擴增和/或檢測。下面的實施例示出了本發明的具體實施方式,但所述實施例不是限制性的。實施例實施例1:使用帶標簽引物的切口thda在該實施例中,使用標準thda和本發明的切口thda方法分別擴增和檢測了來自于淋病奈瑟氏菌基因組的孔蛋白基因的序列(seqidno:1):5’-atttgttccgagtcaaaacagcaagtccgcctatacgcctgctactttcacgctggaaagtaatcagatgaaaccagttccg-3’a)材料:表1:用于擴增和檢測seqidno:1的引物和探針tm:熔融溫度;標準thda:引物poraf5和porar5,探針pora5_vd5fam(標記有6-fam和bhql);切口thda:引物poraf5和pora_ns,探針pora5_vd5fam(標記有6-fam和bhql)表2:用于淋病奈瑟氏菌(n.gonorrhoeae)基因組dna的標準thda和切口thda的使用表1的引物的預混物1反應混合物:2.5μl10x退火緩沖液0.2μl5mnacl0.4μl25mmdntp混合物0.75μll00mmdatp1μl100mmmgso410x退火緩沖液:100mmkcl200mmtris/hclph8.8表3:用于淋病奈瑟氏菌基因組dna的標準thda和切口thda的使用表1和2的引物和試劑的預混物2用于電泳的12%聚丙烯酰胺凝膠:12ml30%丙烯酰胺/雙丙烯酰胺29:13mltbe13mlh2o150μl10%aps40μltemeda)方法:-用移液管將22μl預混物1吸到8xpcr排管中-在pcr循環儀中在95℃下2min的變性步驟,然后立即在4℃下冷卻,-然后將排管在管掃描器(tubescanner)(esegmbh)中在65℃下溫育1min-在蓋上蓋離心后,用移液管向排管的蓋子中各加入3μl預混物2-將反應混合物通過振搖進行混合,再次離心,并置于管掃描器中-然后在65℃下將管溫育60min,每30秒讀取熒光-接著將各10μl的反應物轉移到rotorgene管中,并用rotorgeneq(qiagengmbh)測定熔融曲線(55-95℃,增量0.5℃,保持5")-將各4μl的反應物荷載到12%聚丙烯酰胺凝膠(70min,140v)上c)結果:i)實時(切口)thda:圖4示出了在管掃描器中不同的實時(切口)thda反應的結果(擴增曲線)。圖中所示的是隨時間推移的原始熒光強度。-所有無模板對照反應(ntc)均沒有顯示出非特異性擴增-標準thda的擴增曲線在約15min處具有分接點(take-offpoint)-無nb.bsmi的切口thda的擴增曲線在24min處具有分接點,并具有顯著小于另兩條曲線的斜率-有nb.bsmi的切口thda的擴增曲線在約11min處具有分接點ii)熔融曲線:圖5示出了擴增產物的熔融曲線。a)標準thda,b)無nb.bsmi的切口thda,c)有nb.bsmi的切口thda。對于所有包含靶(模板)dna的反應,通過熔融溫度(tm)均可鑒定到特異性擴增產物。在所有ntc反應中,沒有觀察到可檢測的擴增子。iii)聚丙烯酰胺凝膠:圖6示出了具有擴增和電泳后的各個反應混合物的聚丙烯酰胺凝膠。a)染色前的凝膠,b)用etbr染色后的凝膠。凝膠上各個條帶b1至b5的內含物還被圖示在圖6b1至6b5中。泳道1:淋病奈瑟氏菌gdna的標準thda泳道2:ntc標準thda泳道3:淋病奈瑟氏菌gdna的無nb.bsmi的切口thda泳道4:ntc無nb.bsmi的切口thda泳道5:淋病奈瑟氏菌gdna的有nb.bsmi的切口thda泳道6:ntc有nb.bsmi的切口thdal:10bp梯狀條帶(大小標記物)未染色凝膠上的條帶(圖6a)是由于熒光標記的探針引起的。這些條帶證實了所預期的產物對引物和切口核酸內切酶的初始量的依賴性(b1-b3)。在染色凝膠上,可觀察到dsdna擴增產物(b4,b5)以及與探針的雜交產物(b1-b3)的顯著條帶,所述雜交產物在未染色凝膠上也可被觀察到。所有ntc反應也顯示出條帶,但是所述條帶在管掃描器或tm分析中由于在該檢測模式(ssdna反向鏈與探針的特異性雜交)中它們的非特異性序列,因此均沒有引起假陽性信號。所有ntc反應在未染色凝膠中也沒有顯示出條帶,在未染色凝膠中的條帶指示與探針雜交。實施例2:使用不帶標簽引物的切口thdaa)靶序列靶序列(seqidno:6)源自于人類p53基因的mrna,并包含序列nm001126114的核苷酸殘基322至395。為了設計包含由切口核酸內切酶識別的序列的引物,對反向引物hda-tp53rev的原始序列進行修飾,從而產生了引物序列hda-tp53rev7。這引入了酶nt.bstnbi的識別序列(參見圖7)。表4:用于擴增的引物序列引物id序列id序列5’→3’hda-tp53forseqidno:7atttgatgctgtccccggacgatatthda-tp53revseqidno:8cattctgggagcttcatctggaccgthda-tp53rev7seqidno:9cattctgggagtctcatctggacctgb)從人類cdna擴增p53材料:對于該反應,將人類rna轉錄成cdna。對于thda反應,使用來自于10ngrna的cdna。在表5所概述的緩沖液(反應混合物)中執行該反應。表5:用于thda擴增p53cdna的反應混合物tris,ph8.810mmkcl5mmmgso43.5mmnacl40mmdntp0.4mmdatp3mm甜菜堿975mm引物hda-tp530.1μm引物hda-tp53rev70.1μmnt.bstnbi切口核酸內切酶0-10u/反應bst聚合酶1u/μl解旋酶10ng/μl反應混合物已被置于冰上,然后在實時pcr循環儀中在65℃下執行60min。結果:圖8示出了反應速率對切口核酸內切酶的濃度的依賴性。圖中示出的是在可以檢測到顯著高于背景信號的擴增子信號之前的以分鐘(min)為單位的時間。在存在較低濃度的切口核酸內切酶時,比無切口核酸內切酶的反應早約5min檢測到信號,表示反應更快。圖9示出了標準thda即無任何切口核酸內切酶的thda的擴增曲線(隨時間推移的熒光)。在cdna模板存在下測得三條曲線,在cdna模板不存在下測得兩條曲線。圖10示出了在0.1u切口核酸內切酶nt.bstnbi存在下thda的擴增曲線。在cdna模板存在下測得三條曲線,在cdna模板不存在下測得兩條曲線。圖11示出了在無切口核酸內切酶nt.bstnbi的標準thda之后的熔融曲線分析。對圖9中示出的反應進行熔融曲線分析。在cdna模板存在下測得三條曲線,在cdna模板不存在下測得兩條曲線。圖12示出了在0.1u切口核酸內切酶nt.bstnbi存在下標準thda之后的熔融曲線分析。對圖10中示出的反應進行熔融曲線分析。在cdna模板存在下測得三條曲線,在cdna模板不存在下測得兩條曲線。c)從人類基因組dna(gdna)擴增p53100ng人類基因組dna被用于該反應,并在95℃加熱步驟中進行變性。在表6所概述的反應緩沖液中執行該反應。表6:用于thda擴增p53gdna的反應混合物tris,ph8.810mmkcl5mmmgso43.5mmnacl40mmdntp0.4mmdatp3mm甜菜堿975mm引物hda-tp530.1μm引物hda-tp53rev70.1μmnt.bstnbi切口核酸內切酶0-0.4u/μlbst聚合酶1u/μl解旋酶10ng/μl反應混合物已被置于冰上,然后在實時pcr循環儀中在65℃下執行60min。結果:圖13示出了反應速率對切口核酸內切酶的存在的依賴性。圖中示出的是在可以檢測到顯著高于背景信號的擴增子信號之前的以分鐘(min)為單位的時間。在切口核酸內切酶存在下,比無切口核酸內切酶的反應早約6min檢測到信號,表示反應更快。序列表<110>凱杰有限公司<120>使用切口酶的解旋酶依賴性等溫擴增<130>s2336pctbln<150>ep10173094.3<151>2011-08-10<160>9<170>patentinversion3.5<210>1<211>82<212>dna<213>neisseriagonorrhoeae<400>1atttgttccgagtcaaaacagcaagtccgcctatacgcctgctactttcacgctggaaag60taatcagatgaaaccagttccg82<210>2<211>27<212>dna<213>artificialsequence<220><223>primer/probe<400>2atttgttccgagtcaaaacagcaagtc27<210>3<211>27<212>dna<213>artificialsequence<220><223>primer/probe<400>3cggaactggtttcatctgattactttc27<210>4<211>25<212>dna<213>artificialsequence<220><223>primer/probe<400>4cgcctatacgcctgctactttcacg25<210>5<211>50<212>dna<213>artificialsequence<220><223>primer/probe<400>5cggggcgtctggaaggcgcattccggaactggtttcatctgattactttc50<210>6<211>74<212>dna<213>homosapiens<400>6atttgatgctgtccccggacgatattgaacaatggttcactgaagacccaggtccagatg60aagctcccagaatg74<210>7<211>26<212>dna<213>artificialsequence<220><223>primer/probe<400>7atttgatgctgtccccggacgatatt26<210>8<211>26<212>dna<213>artificialsequence<220><223>primer/probe<400>8cattctgggagcttcatctggaccgt26<210>9<211>26<212>dna<213>artificialsequence<220><223>primer/probe<400>9cattctgggagtctcatctggacctg26當前第1頁12
相關技術
網友詢問留言
已有0條留言
- 還沒有人留言評論。精彩留言會獲得點贊!
1
- 環介導等溫擴增多重耐藥基因oqxAB的檢測引物、檢測試劑盒及檢測方法與制造工藝
- 一種氣單胞菌的核酸等溫擴增檢測試劑盒及其檢測方法與制造工藝
- 一種霍亂弧菌環介導等溫擴增試劑盒及其應用的制造方法與工藝
- 用于檢測番茄晚疫病菌的LAMP引物組合物和應用的制造方法與工藝
- 基于顏色判定的環介導等溫擴增(LAMP)技術檢測雪松疫霉根腐病菌的制造方法與工藝
- Sortase A介導的(S)?羰基還原酶寡聚體高效催化(S)?苯基乙二醇的合成的制造方法與工藝
- 用于旋轉機器中的超導線圈的等溫支承部和真空容器的制造方法與工藝
- 用于擴增細胞群的方法、試劑盒及裝置與制造工藝
- 基于環介導等溫擴增消光技術的轉基因玉米MIR604檢測方法與制造工藝
- 基于環介導等溫擴增消光技術的玉米檢測方法與制造工藝
- 一種霍亂弧菌環介導等溫擴增試劑盒及其應用的制造方法與工藝
- 用于檢測番茄晚疫病菌的LAMP引物組合物和應用的制造方法與工藝
- 基于顏色判定的環介導等溫擴增(LAMP)技術檢測雪松疫霉根腐病菌的制造方法與工藝
- Sortase A介導的(S)?羰基還原酶寡聚體高效催化(S)?苯基乙二醇的合成的制造方法與工藝
- 用于旋轉機器中的超導線圈的等溫支承部和真空容器的制造方法與工藝
- 基于環介導等溫擴增消光技術的轉基因玉米MIR604檢測方法與制造工藝
- 基于環介導等溫擴增消光技術的玉米檢測方法與制造工藝
- 一套鑒定甘蔗上三種短體線蟲的環介導等溫擴增引物及其試劑盒的制造方法與工藝
- RNA擴增方法與制造工藝
- 用于擴增核酸樣品的等溫方法與制造工藝