一種雙核鎂?鍺鎢氧簇催化劑、制備方法及其用途與流程
本發明屬于多金屬氧簇催化劑材料制備技術領域,其中涉及到一種雙核鎂-鍺鎢氧簇催化劑的制備技術,為其催化應用奠定基礎。
技術背景
多金屬氧簇化學發展至今已經歷了近兩百多年的歷史,已經成為當前無機化學中發展最快、交叉最廣的領域之一。早期的多金屬氧簇化學認為無機含氧酸(如磷酸、硫酸、鎢酸、鉬酸等)經縮合可形成縮合酸。多金屬氧簇化學就是關于同多酸和雜多酸的化學,前者涉及同多陰離子,后者涉及雜多陰離子。在多金屬氧簇中,鎢能形成許多的同多及雜多鎢氧簇合物。實驗表明,該類化合物在催化、相轉移、選擇性氧化及藥物化學方面具有較好的前景。
硫醚氧化為相應的砜類化合物是有機合成及工業生產的重要反應。究其原因是因為硫醚氧化物亞砜和砜具有良好的生物活性,在農藥、醫藥等精細化學品中得到廣泛的應用。重要的是,砜和亞砜是有機合成反應中重要的合成中間體,在合成功能材料和分子重組等反應中具有廣闊的應用前景。其主要用作制造染料、醫藥、香料、調味品、農藥等重要中間體的原料。在硫醚氧化反應中,H2O2作為綠色氧化劑而備受關注,可是H2O2作為氧化劑存在的問題是其氧化能力中等。因此,為了較好地以H2O2作為氧化劑實現綠色氧化硫醚化合物的主要問題是建立高效、高選擇性的催化體系。因此選擇合適的催化劑成為解決這一問題的關鍵。
近年來,文獻報道中有機無機雜化的鎢磷氧簇(Ezzat Rafie, et al, Journal of Molecular Catalysis A: Chemical, 2013,380, 18-27)氧化苯硫醚,但是在以上反應中存在著催化劑合成較為復雜等問題。
基于以上文獻的基礎上,設計一個硫醚氧化生成砜、合成簡單的高效催化體系是十分必要的。通過檢索,尚未發現與本發明申請相關的公開專利文獻。
技術實現要素:
本項發明的目的是解決以往催化劑在雙氧水為氧化劑下硫醚的選擇性氧化過程中合成較為復雜等問題。提供了一種合成新型雙核鎂-鍺鎢氧簇催化劑的制備方法,以期在硫醚的選擇性氧化中具有較好的催化作用,實現硫醚分子選擇性氧化的目標。
本發明的方案是,雙核鎂-鍺鎢氧簇催化劑,其特征是,結構式為:
雙核鎂-鍺鎢氧簇催化劑的制備方法,將金屬鎂鹽、DMF在水溶液中與鍺鎢酸反應構筑金屬雙核鎂-鍺鎢氧簇;利用自然揮發法得到雙核鎂-鍺鎢氧簇催化劑的單晶。
所述的制備方法,優選的是,在一個潔凈的燒杯中依次加入氯化鎂、N,N’-二甲基甲酰胺(DMF)溶解在水中, 攪拌溶解,再加入鍺鎢酸,加熱攪拌,過濾,濾液在室溫下緩慢蒸發,4~6天后得到無色晶體,產率約47~62%。
所述的制備方法,優選的是,在30~50℃下攪拌6~10h(優選的,在40℃下攪拌8 h)。
所述的制備方法,優選的是,氯化鎂:N,N’-二甲基甲酰胺:鍺鎢酸物質的量之比為2~4 : 15~30 : 1~3。
本發明還提供了所述鍶-鍺鎢氧簇催化劑在硫醚選擇性氧化中的用途。硫醚為苯甲硫醚、對氯苯甲硫醚、對硝基苯甲硫醚、對甲氧基苯甲硫醚等。
本發明的設計思路如下:
將金屬鎂鹽與DMF在水溶液中與鍺鎢酸發生取代反應構筑雙核鎂-鍺鎢氧簇配合物;并利用自然揮發法得到了雙核鎂-鍺鎢氧簇催化劑的單晶;
將具有明確結構的雙核鎂-鍺鎢氧簇催化劑應用于硫醚的雙氧水條件下選擇性催化氧化中,實現硫醚選擇性氧化的高轉化率高選擇性的目標。
這類催化劑的晶體結構信息通過如下方法獲得的:
通過常規的溶液反應合成得到雙核鎂-鍺鎢氧簇催化劑的晶體,具體的描述實驗方法如下:
在100mL的燒杯中依次加入氯化鎂(2 ~4mmol)、DMF(15 ~30mmol)以及水 50 ~80 mL, 攪拌溶解,再加入鍺鎢酸(1 ~3mmol),在30~50 ℃下攪拌6~10h,冷卻至室溫,過濾,濾液在室溫下緩慢蒸發,4~6天后得到無色晶體[Mg(HCON(CH3)6]2GeW12O40。產率約47~59%。
產品通過單晶X衍射,元素分析進行表征,得到關于晶體結構的準確信息。具體的結果如下:
晶體的分子式為[Mg(HCON(CH3)6]2GeW12O40, 其中陽離子部分為金屬鎂與DMF形成的配合陽離子,陰離子為GeW12O40,二者通過靜電引力相互作用結合在一起。
這項發明主要是合成雙核鎂-鍺鎢氧簇催化劑,已經將其應用于硫醚類化合物的選擇性氧化。這類催化劑可以實現硫醚化合物的選擇性氧化,轉化率高達92.7%,選擇性高達94.9%。該類催化劑的制備方法反應過程簡單。
上述硫醚為苯甲硫醚、對氯苯甲硫醚、對甲基苯甲硫醚、對甲氧基苯甲硫醚等,轉化率,選擇性通過氣相色譜檢測。
本發明提供雙核鎂鎢氧簇催化劑具有以下特點:
制備方法簡單、并且催化劑都具有明確的分子結構,有利于研究催化反應機理。
催化劑便于容易分離,經處理后可以多次使用,并且仍能保持良好的催化活性,有利于工業化生產。
附圖說明
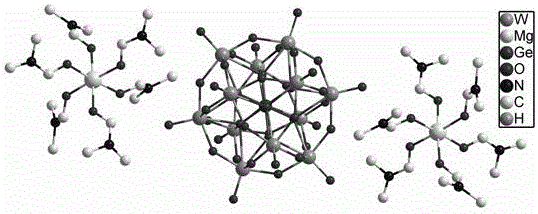
圖1. 化合物[Mg(HCON(CH3)6]2GeW12O40的晶體結構。
具體實施方式
下面結合實施例對本發明作詳細描述,但保護范圍不被此限制。
本發明所用原料皆可從市場購買,比如DMF全稱N,N’-二甲基甲酰胺,購自國藥集團化學試劑有限公司。
實施例1:化合物[[Mg(HCON(CH3)6]2GeW12O40的制備
在100mL的燒杯中依次加入氯化鎂(2mmol)、DMF(16mmol)以及水50mL, 攪拌溶解,再加入鍺鎢酸(1mmol),在40℃下攪拌6h,冷卻至室溫,過濾,濾液在室溫下緩慢蒸發,4~6天后得到無色晶體[Mg(HCON(CH3)6]2GeW12O40。產率約56%。
實施例2:化合物[[Mg(HCON(CH3)6]2GeW12O40的制備
在100mL的燒杯中依次加入氯化鎂(3mmol)、DMF(30mmol)以及水50 mL, 攪拌溶解,再加入鍺鎢酸(2mmol),在50℃下攪拌10h,冷卻至室溫,過濾,濾液在室溫下緩慢蒸發,4~6天后得到無色晶體[Mg(HCON(CH3)6]2GeW12O40。產率約52%。
實施例3:化合物[[Mg(HCON(CH3)6]2GeW12O40的制備
在100mL的燒杯中依次加入氯化鎂(4mmol)、DMF(15mmol)以及水80 mL, 攪拌溶解,再加入鍺鎢酸(3mmol),在30℃下攪拌6h,冷卻至室溫,過濾,濾液在室溫下緩慢蒸發,4~6天后得到無色晶體[Mg(HCON(CH3)6]2GeW12O40。產率約49%。
實施例4:化合物[[Mg(HCON(CH3)6]2GeW12O40的制備
在100mL的燒杯中依次加入氯化鎂(4mmol)、DMF(30mmol)以及水50mL, 攪拌溶解,再加入鍺鎢酸(1mmol),在30℃下攪拌8h,冷卻至室溫,過濾,濾液在室溫下緩慢蒸發,4~6天后得到無色晶體[Mg(HCON(CH3)6]2GeW12O40。產率約47%。
實施例5:化合物[[Mg(HCON(CH3)6]2GeW12O40的制備
在100mL的燒杯中依次加入氯化鎂(3mmol)、DMF(20mmol)以及水70 mL, 攪拌溶解,再加入鍺鎢酸(2mmol),在50 ℃下攪拌10h,冷卻至室溫,過濾,濾液在室溫下緩慢蒸發,4~6天后得到無色晶體[Mg(HCON(CH3)6]2GeW12O40。產率約61%。
實施例6:化合物[[Mg(HCON(CH3)6]2GeW12O40的制備
在100mL的燒杯中依次加入氯化鎂(4mmol)、DMF(30mmol)以及水50mL, 攪拌溶解,再加入鍺鎢酸(2mmol),在40 ℃下攪拌8h,冷卻至室溫,過濾,濾液在室溫下緩慢蒸發,4~6天后得到無色晶體[Mg(HCON(CH3)6]2GeW12O40。產率約55%。
實施例7:化合物[[Mg(HCON(CH3)6]2GeW12O40的制備
在100mL的燒杯中依次加入氯化鎂(2mmol)、DMF(15mmol)以及水60mL, 攪拌溶解,再加入鍺鎢酸(3mmol),在30℃下攪拌6h,冷卻至室溫,過濾,濾液在室溫下緩慢蒸發,4~6天后得到無色晶體[Mg(HCON(CH3)6]2GeW12O40。產率約51%。
實施例8:化合物[[Mg(HCON(CH3)6]2GeW12O40的制備
在100mL的燒杯中依次加入氯化鎂(2mmol)、DMF(30mmol)以及水70 mL, 攪拌溶解,再加入鍺鎢酸(2mmol),在40℃下攪拌8h,冷卻至室溫,過濾,濾液在室溫下緩慢蒸發,4~6天后得到無色晶體[Mg(HCON(CH3)6]2GeW12O40。產率約58%。
實施例9:化合物[[Mg(HCON(CH3)6]2GeW12O40多金屬氧簇催化劑對硫醚類化合物的催化氧化應用
以化合物[[Mg(HCON(CH3)6]2GeW12O40為例:取0.25mmol硫醚溶于1 mL甲醇中,0.3mmol雙氧水,加入催化劑20mg, 40 °C下反應4-8小時,用氣相色譜檢測,具體硫醚氧化的數據見表2.
表2. 化合物[Mg(HCON(CH3)6]2GeW12O40 對硫醚選擇性氧化結果列表
圖1為化合物[[Mg(HCON(CH3)6]2GeW12O40的晶體結構。
表1為該化合物的晶體學數據。
表1
- 還沒有人留言評論。精彩留言會獲得點贊!
- 一種高性能抗指紋劑的制備方法
- 一種常壓下光催化合成苯胺類化合物的方法及其使用的催化劑的制作方法
- 一種底物按次序催化的金屬納米粒子基催化劑的制作方法
- 一種Cu?ZnO?ZrO<sub>2</sub>?NH<sub>2</sub>/SBA?15復合催化劑的制備方法及其應用
- 一種催化氣化灰渣中不可溶性堿金屬催化劑的回收方法
- 一種催化氧化丙烷制取丙烯酸的催化劑及其制備方法
- 催化氧化丙烷制取丙烯酸的催化劑及其制備方法
- 一種催化合成γ-氯丙基三氯硅烷的方法及其催化劑的制作方法
- 一種高性能氧還原MnO<sub>2</sub>-Mn<sub>3</sub>O<sub>4</sub>/碳納米管復合催化劑及其制備方法和應用
- 錫類催化劑催化合成聚乳酸共聚改性產物的方法