親水性藥物的靶向遞送的制作方法
發明領域本發明涉及將親水性藥物給藥到人體或動物的肝臟,以及給藥到實體肝臟瘤。更具體來說,本發明涉及親水性藥物,特別是抗癌藥物的給藥。
背景技術:
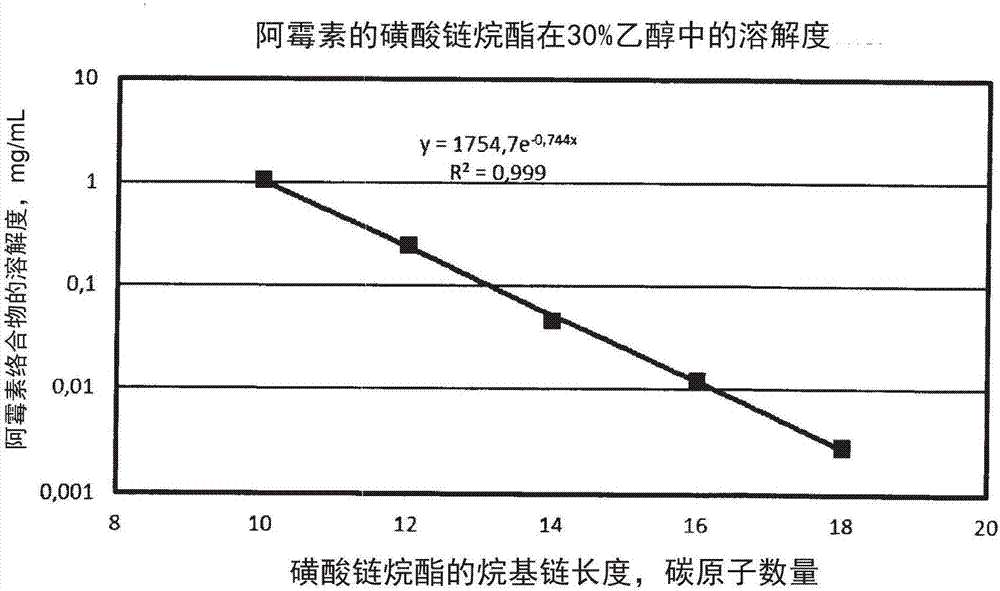
:治療窗口表示可以對疾病進行有效和安全治療的藥物劑量范圍。其包括觀察到明顯治療效果的劑量至治療益處被不利影響中和的劑量。大部分的抗癌藥物具有窄治療窗口。此外,通常很少比例的給藥劑量到達待治療位點。通過口服攝取或血管注射進行系統給藥之后,藥物經由循環分布在整個人體,導致整個人體受到影響。理想地,藥物應該被專門導向到所需的體部位,例如需要進行治療的器官或組織。該靶向給藥會避免對身體的其余部分造成傷害。這種給藥類型需求將藥物導向到感興趣的組織,同時避免大量藥物抵達不需要治療的組織。需要被導向到特定身體部位的藥物例子是抗癌藥物阿霉素。通常接受的是,通過靶向藥物遞送,可以明顯改善阿霉素的治療潛力,因為由此可以避免或者至少明顯降低其有害副作用。阿霉素的最危險副作用是對心臟的損害。當阿霉素的累積劑量達到550mg/m2時,發生心臟副作用的風險急劇增加。阿霉素心臟中毒表征為線粒體氧化磷酸化中的劑量依賴性下降。由于阿霉素與鐵相互作用產生的反應性氧物質會損傷肌細胞,導致肌原纖損耗和細胞質空泡形成。此類過度損傷可能導致患者死亡。因此,希望保持阿霉素的心臟濃度盡可能的低。肝癌是需要給藥阿霉素的許多惡性腫瘤之一。更具體來說,可以通過稱作經動脈化療栓塞的工藝進行靶向(或局部)給藥來治療肝癌。在該過程中,將阿霉素通過導管直接給藥到動脈,給藥到肝臟的患病部分,同時通過例如海綿膠堵住未受到疾病影響的肝臟的動脈供給部分。隔離肝臟輸注是用于肝臟的藥物靶向遞送的一種方法。其包括外科手術,在該過程中,從體循環隔離到達肝臟的血液循環,并用血液分開輸注。在隔離之后,通過輸注到肝臟的血液注入藥物,實現較高濃度的化學治療試劑用于系統給藥。但是,該侵入式過程在技術上是復雜且不安全的。許多藥理活性劑,例如前述的抗癌藥是包含一個或多個氨基的弱堿。由于該原因,它們與強酸和弱酸形成鹽,并且通常以鹽形式給藥。它們的常見的藥學可接受鹽(特別是它們的鹽酸鹽、氫溴化物、磷酸鹽、硫酸鹽、乳酸鹽、酒石酸鹽等)在水性體流體中的溶解度通常高于游離堿的溶解度。因此,此類鹽的水性溶液而不是相應堿的水性溶液被用于靜脈注射。為了給藥到人體或動物肝臟,此類抗癌藥以陽離子兩親形式提供(藥學可接受酸的鹽形式)。這種給藥方式應用于抗癌藥,例如,蒽環霉素(阿霉素、表柔比星、柔紅霉素、伊達比星、米托蒽醌),長春花屬生物堿(長春堿、長春新堿、長春瑞濱),安吖啶,拓撲替康和伊立替康,但不限于此。如果藥學活性劑包含不止一個氨基,則它們中的兩個或更多個可以質子化并與酸形成鹽。前述類型的陽離子兩親藥物(cad)與兩親陰離子表面活性劑(例如,磺酸鏈烷酯)反應,形成不溶于水的絡合物。雖然仍然符合有機堿與有機酸或無機酸的鹽的定義,但是不溶于水的絡合物在一定程度上額外通過非共價鍵作用力連接。發明目的本發明的一個主要目的是提供用于肝臟抗癌藥物的靶向給藥的藥學組合物,其包含一個或多個氨基官能團,其不含已知藥物組合物的一種或多種缺陷或者至少在一定程度上展現出對于這些缺陷的減少。本發明的另一個目的是提供用于實體肝臟瘤抗癌藥物的靶向給藥的藥學組合物,其包含一個或多個氨基官能團,其不含本領域已知藥物組合物的一種或多種已知缺陷或者至少在一定程度上展現出對于這些缺陷的減少。本發明的另一個目的是提供一種此類藥學組合物的設計方法,其在靜脈給藥之后,會在肝臟或實體肝臟瘤中提供所需的靶向藥物濃度。通過研究本發明的如下簡要描述、其優選實施方式以及所附權利要求,本發明的其他目的會是顯而易見的。技術實現要素:本發明基于如下理解:兩親化合物(特別是包含氨官能團的親水性抗癌藥物的磺酸直鏈烷基酯和硫酸直鏈鏈烷酯)顆粒的水性懸液是有價值的形式,通過這種形式,可以將這些藥物以集中其治療效果的方式給藥到肝臟或實體肝臟瘤,即,肝臟或實體瘤的靶向定位。因此,靶向定位理解為優先遞送到肝臟從而在肝臟組織(包括肝癌組織)中實現相比于其他組織更高的藥物濃度(w/w)。親水性抗癌藥物的磺酸直鏈烷基酯和硫酸直鏈鏈烷酯的一個重要特征在于它們在水和水性體液中在25℃時小于0.1mg/ml的低溶解度。作為本發明背后的一種可能的生物學解釋(但不以任何方式約束本發明)是,在向系統循環或肝臟或實體肝臟瘤給藥了此類抗癌藥物的顆粒水性懸液之后,藥物顆粒會在給定時間段內在體內達到平衡分布。它們在水性介質中的溶解度是非常低的,但并非為零。因此,它們會緩慢溶解在體液中,直至達到由它們的溶解度決定的平衡。由于溶解的材料不可逆地化學轉變成降解產物,隨時間溶解了更多的材料,從而維持平衡。只要通過材料溶解供給了平衡,就局部地維持了藥物的穩態濃度。本發明還基于如下理解:包含氨官能團的親水性抗癌藥物的兩親磺酸直鏈烷基鹽/酯的水性膠體以及包含氨官能團的親水性抗癌藥物的兩親硫酸直鏈鏈烷鹽/酯的水性膠體是有價值的形式,通過這種形式,可以將這些藥物以集中其治療效果的方式給藥到所需器官(特別是肝臟),或者給藥到所需實體瘤(即,器官或實體瘤的靶向定位)。水性膠體由粒度最高至約10000nm的顆粒構成,或者包括粒度最高至約10000nm的顆粒。本發明的水性膠體的一個重要性質在于它們的低沉淀速率。通常來說,隨著粒度的增加,給定顆粒種類的沉淀速率增加。但是,通過增加水性相的粘度和/或通過改變顆粒的表面性質(例如,表面粗糙度),可以防止其發生增加并且甚至可以使其下降。本發明提供了親水性癌癥藥物的鹽的固體顆粒,其包含一個或多個氨基以及水溶性硫酸烷基鹽/酯或磺酸鏈烷鹽/酯或者兩種或更多種此類硫酸鹽/酯或磺酸鹽/酯的混合物。鹽的一個重要特征在于其在水中的低溶解度。換言之,本發明的鹽是基本不可溶的。“基本不可溶”應理解為在水中或水性體液中的溶解度小于0.1重量%,特別地,小于0.05或0.02重量%。本發明提供了具有水溶性硫酸烷基酯或磺酸鏈烷酯或者具有此類硫酸酯或磺酸酯中的兩種或更多種的混合物的親水性癌癥藥物的不溶于水的鹽的固體顆粒的生產方法。本發明還提供了藥學組合物,其包含一種或多種本發明的兩親磺酸鹽/酯和/或硫酸鹽/酯以及液體載劑。可以通過任意合適途徑,例如動脈內、腹膜內、肌肉內、透皮或靜脈給藥方式,來給藥該組合物。優選以一劑包含本發明的兩親磺酸鹽/酯和硫酸鹽/酯的水性膠體的進行給藥。本發明還提供了包含親水性癌癥藥物的水不溶性鹽和水溶性硫酸烷基酯或磺酸鏈烷酯,或者兩種或更多種此類硫酸鹽或磺酸鹽的混合物的固體顆粒形式的藥物組合物的生產方法。本發明的組合物還可包含緩沖劑和藥學可接受賦形劑,例如滲透壓(osmolality)控制劑和粘度控制劑。由于采用的生產方法,組合物額外含有由水溶性硫酸烷基酯或磺酸鏈烷酯的陽離子以及抗癌藥物的陰離子構成的鹽或對應離子。優選使用堿性硫酸烷基酯和堿性磺酸鏈烷酯,特別是硫酸烷基鈉和硫酸烷基鉀以及磺酸鏈烷鈉和磺酸鏈烷鉀。本發明的兩親顆粒磺酸鹽/酯和硫酸鹽/酯由藥理學試劑d和一種或多種硫酸酯或磺酸酯陰離子構成,所述藥理學試劑d具有抗癌活性,其包含1-4個氨基(其中的一個或多個被質子化)。它們表示為如下化學式(1)和(2):dn+(r1so3)-n(1)dn+(r2oso3)-n(2)其中,r1是直鏈c6-c30烷基;r2是直鏈c6-c30烷基;n是1-4的整數。r1和r2優選是直鏈c10-c20烷基,更優選是直鏈c12-c18烷基,甚至更優選約為直鏈c16烷基。因此,r1可以是直鏈c12、c13、c14、c15、c16、c17、c18烷基中的任意一種;r2是直鏈c12、c13、c14、c15、c16、c17、c18烷基中的任意一種。90%的膠體顆粒的優選粒度小于或等于10000nm,更優選小于或等于5000nm。根據本發明的一個優選方面,本發明包括尺寸大于膠體顆粒的顆粒以及它們的水性懸液,例如,粒度最高至10μm或50μm以及甚至最高至100μm,以及它們的懸液。可以通過例如,離心或冷沉淀,從水性相分離本發明的顆粒。如果通過離心進行分離,則水性相消除了伴隨而來的由水溶性硫酸烷基酯或磺酸鏈烷酯的陽離子以及抗癌藥物的陰離子構成的鹽或對應離子。所得到的粉末(如果需要的話,進行額外干燥)保留至少50%,更優選至少80%的膠體粒度。為了促進在水性介質中的再懸浮,粉末可以包含再懸浮促進劑,例如葡萄糖、乳糖或白蛋白。或者,可以通過水性介質的蒸發(包括冷沉淀)生產本發明的顆粒;以這種方式,它們會與伴隨而來的鹽混合,所述鹽包含水溶性硫酸烷基酯或磺酸鏈烷酯的陽離子和抗癌藥物的陰離子;如果需要的話,它們可以與再懸浮促進劑混合。根據本發明的另一個優選方面,本發明的膠體顆粒可以包含對于待治療的腫瘤的表面抗原具有親和力的微載體顆粒,例如通過包含合適的抗體結構。本發明的兩親磺酸鹽/酯和硫酸鹽/酯的優選藥理學活性劑d包括但不限于:阿霉素、表柔比星、柔紅霉素、伊達比星(idarubicin)、米托蒽醌、長春堿、長春新堿、長春瑞濱、安吖啶、拓撲替康、伊立替康。根據本發明的另一個優選方面,除了上文所述的那些,用于制備本發明的兩親磺酸鹽/酯和硫酸鹽/酯的合適的抗癌劑是具有與阿霉素、表柔比星、柔紅霉素、伊達比星、米托蒽醌、長春堿、長春新堿、長春瑞濱、安吖啶、拓撲替康、伊立替康類似的親水性。根據本發明,還揭示了治療人體中的肝癌的方法,其包括:向所述人體給藥治療有效量的本發明的藥物組合物或者包含本發明的兩親顆粒磺酸鹽/酯或硫酸鹽/酯粉末的藥物組合物。一種優選的給藥方法是通過輸注或注射進入靜脈或動脈,特別是進入門靜脈或肝動脈。另一種優選的給藥方法是通過輸注或注射進入外周循環到達實體肝臟瘤。第三種優選的給藥方法是直接輸注或注射進入實體肝臟瘤。根據本發明的一個優選方面,優選通過一劑或者多劑給藥。向肝臟或肝臟瘤給藥的一個方面是通過輸注或注射進入靜脈或動脈,特別是進入門靜脈或肝動脈。根據本發明,提供了用于方便和非侵入式給藥的藥物遞送系統,其能夠在延長的時間段上(例如,超過1小時或6小時或者甚至大于或等于1天)提供所需的藥物濃度。在肝臟或者實體肝臟瘤或者其他組織中提供并維持了此類濃度。根據本發明的一個優選方面,本發明提供了控制肝臟與其他器官和組織之間的藥物分布比的方法。在本申請中,除非另有說明,否則術語“肝臟疾病”(肝病)包括原發性肝癌(例如,肝細胞癌和/或膽管癌、血管肉瘤、肝的血管肉瘤),繼發性惡性腫瘤(即,從原發性癌癥轉移的胃腸道和其他器官(例如腎、肺、乳腺或前列腺)中的繼發性損傷);由于病毒引起的肝臟感染(病毒性肝炎)、肝毒素(例如酒精性肝炎)、自身免疫性(自身免疫性肝炎)或遺傳性病癥;過度飲酒引起的肝硬化。根據本發明,還揭示了藥物組合物的設計方法,其用于在預定時間段內,為選自肝臟或實體肝臟瘤的人或動物的治療靶標提供預定濃度的藥理學活性劑d的硫酸鹽/酯或磺酸鹽/酯(其包含1-4個如下化學式(1)或(2)表示的氨基)或者這些試劑的混合物:dn+(r1so3)-n(1)dn+(r2oso3)-n(2)其中,r1是直鏈c6-c30烷基;r2是直鏈c6-c30烷基;n是1-4的整數;其中,該方法包括:i)對于各種碳鏈長度x、y,確定dn+(r1so3-)n和/或dn+(r2oso3-)n在水性溶劑中的溶解度;ii)在將所述藥理學活性劑d給藥到對象或動物之后,確定所述藥理學活性劑的所述硫酸鹽/酯或磺酸鹽/酯的溶解度與所述藥理學活性劑d在治療靶中的預期濃度之間的相關性;iii)基于所述藥理學活性物質d在所述器官或組織中的所需濃度,限定所述藥理學活性劑的所述硫酸鹽/酯或磺酸鹽/酯在所述溶劑中的目標溶解度;iv)確定對應所述目標溶解度的碳鏈長度x、y;v)提供包含由此確定的碳鏈長度x、y的所述藥理學活性劑的硫酸鹽/酯或磺酸鹽/酯;vi)提供流體載劑;vii)將包含由此確定的碳鏈長度x、y的所述藥理學活性劑的硫酸鹽/酯或磺酸鹽/酯與流體載劑組合,組合的量能夠在所述時間段內維持所述濃度。根據本發明的一個優選方面,確定水性有機溶劑(例如,水性醇,特別是濃度5-50%,優選10-30%(v/v)的水性乙醇)中的溶解度。也可使用其他與水易混溶的溶劑,例如,低分子量酮、酰胺、酯、酰胺和亞砜。根據本發明的一個優選方面,藥物組合物包含:化學式(1)或(2)表示的本發明的至少兩種不同硫酸鹽/酯或磺酸鹽/酯的混合物,或者至少兩種不同的硫酸鹽/酯或磺酸鹽/酯(其中一個由化學式(1)表示,另一個由化學式(2)表示)的混合物。優選的藥理學活性物質d選自下組:阿霉素、表柔比星、柔紅霉素、伊達比星、米托蒽醌、長春堿、長春新堿、長春瑞濱、安吖啶、拓撲替康、伊立替康。還優選組合物包括膠體。優選的流體載劑是水或水性介質,在其中,所述藥理學活性劑d的硫酸鹽/酯或磺酸鹽/酯是不可溶或者基本不可溶的。“基本不可溶”應理解為溶解度小于0.1重量%,特別地,小于0.05或0.02重量%。可以將組合物設計成用于動脈內、腹膜內、肌肉內、經皮或靜脈給藥。可以以任意合適的順序進行上述步驟。所述藥理學活性劑d的所述硫酸鹽/酯或磺酸鹽/酯的一種優選形式是平均粒度(n)最高至100μm、優選最高至50μm或10μm或5μm的粉末。所述藥物組合物的優選形式是水性懸液,特別是水性膠體。根據本發明,還揭示了本發明的藥物組合物的生產方法,該方法包括:提供所述抗癌藥物與非兩親的無機或有機酸的鹽的第一水性溶液;提供第二水性溶液,所述第二水性溶液包含的化學式為(na或k)+(r1so3)-的磺酸烷基酯的鈉鹽或鉀鹽或者化學式為(na或k)+(r2oso3)-的硫酸鏈烷酯的鈉鹽或鉀鹽的量等于所述鹽的量;混合所述第一和第二溶液。雖然可以在該方法中使用除了鈉和鉀之外的鹽,但是使用它們不是優選的。優選地,r1是直鏈c6-c30烷基;r2是直鏈c6-c30烷基;n是1-4的整數。r1和r2優選是直鏈c10-c20烷基,更優選是直鏈c12-c18烷基,最優選約為直鏈c16烷基。根據本發明,還揭示了包含抗癌試劑的本發明的組合物用于靶向遞送到人體或動物的肝臟,所述抗癌試劑選自下組:阿霉素、表柔比星、柔紅霉素、伊達比星、米托蒽醌、長春堿、長春新堿、長春瑞濱、安吖啶、拓撲替康、伊立替康。根據本發明,還揭示了包含抗癌試劑的本發明的兩親顆粒磺酸鹽/酯粉末用于制造包含能夠靶向遞送到人體或動物的肝臟的藥物組合物的藥劑,所述抗癌試劑選自下組:阿霉素、表柔比星、柔紅霉素、伊達比星、米托蒽醌、長春堿、長春新堿、長春瑞濱、安吖啶、拓撲替康、伊立替康。下面將通過本發明的多個非限制性實施例來更詳細闡述本發明。附圖簡要說明圖1顯示阿霉素硫酸烷基酯在30%水性乙醇中的溶解度對于烷基鏈長的依賴性;圖2顯示阿霉素磺酸鏈烷酯在30%水性乙醇中的溶解度對于烷基鏈長的依賴性;圖3顯示米托蒽醌硫酸烷基酯在30%水性乙醇中的溶解度對于烷基鏈長的依賴性;圖4顯示米托蒽醌磺酸鏈烷酯在30%水性乙醇中的溶解度對于烷基鏈長的依賴性;圖5顯示伊立替康磺酸鏈烷酯在10%水性乙醇中的溶解度對于烷基鏈長的依賴性;圖6顯示長春瑞濱磺酸鏈烷酯在20%水性乙醇中的溶解度對于烷基鏈長的依賴性;圖7顯示wistar大鼠注入一劑2小時之后的肝臟中阿霉素濃度對于阿霉素鏈烷硫酸酯和磺酸酯在30%水性乙醇中的溶解度的依賴性;圖8顯示阿霉素的非共價鍵合絡合物在30%水性乙醇中的溶解度與加州兔肝臟靜脈注射0.5小時之后的阿霉素濃度增加之間的關系;圖9顯示在靜脈注射5mg/kg水性阿霉素鹽酸鹽和1.25mg/kg的根據實施例1的本發明的組合物之后的肝臟和心臟中的阿霉素濃度。(圖1-6的)指數趨勢線以及(圖7和8)的對數趨勢線以及它們的等式是通過微軟excel軟件的方式獲得的。具體實施方式一般過程:材料和方法通過如下方式確定在水性乙醇中的溶解度:將充分量的新鮮獲得的膠體以3000rpm離心30-90分鐘,潷析上清液,添加10ml的水并振蕩混合物,然后重復離心,振蕩和清洗3次。來自最終離心的離心液在室溫下空氣干燥72小時,之后真空干燥24小時。一部分的經過干燥的離心液(20mg)通過攪拌再懸浮于6ml的水性乙醇中,處于室溫下持續24小時。混合物在3000rpm離心10分鐘,以及上清液過濾通過0.2微米過濾器以去除未溶解固體產物的團聚物。通過uv法確定化合物的溶解度。用于體內研究的組合物是新鮮制備或者通過濃縮物的稀釋獲得。對于體內研究,選擇年齡為60-75天、體重為300g±30g的雌性wistar大鼠。使用4個動物用于測試一種組合物。以5mg/kg阿霉素的量,通過單劑注入尾部給藥阿霉素組合物。在死亡之后,立即將大鼠體深凍入液氮中。確定阿霉素在肝臟組織中的生物分布。從肝臟的不同部分,選取5或6片總重約為1g的肝臟組織。樣品用含有hcl的水性乙醇溶液以7000rpm均質化20s,以及以11000rpm均質化10s。均化物渦旋30分鐘,以及以3000rpm離心30分鐘。用一氯乙酸溶液處理上清液并孵育1小時。混合物以15000rpm離心15分鐘。通過熒光分析確定最終上清液中的阿霉素濃度。實施例1:制備膠體阿霉素硫酸烷基酯和磺酸鏈烷酯在室溫下,向錐形燒瓶中的5%水性右旋糖中的阿霉素鹽酸鹽溶液(50ml,1mg/ml)添加與阿霉素鹽酸鹽相同的溶劑中的5-10摩爾%過量的na+(r1so3)-或na+(r2oso3)-的溶液。作為5%水性右旋糖的替代,可以在該實施例和其他實施例中使用林格氏溶液或者0.9%的鹽水或磷酸鹽緩沖鹽水或者270-300mosm/l的克分子滲透壓重量濃度的其他水性溶液。目測監測膠體形成過程。在完成添加之后,混合物渦旋或振蕩從30分鐘到7天的額外時間段。然后膠體直接使用或者存儲在冰箱中。通過uv法,以495或233nm確定組合物中阿霉素的濃度。對于取樣,用甲醇稀釋膠體的等分試樣(甲醇過量>20:1)。實施例2:制備膠體米托蒽醌硫酸烷基酯和磺酸鏈烷酯在室溫下,向錐形燒瓶中的5%右旋糖水溶液中的米托蒽醌二鹽酸鹽的劇烈攪拌溶液(40ml,0.2mg/ml)添加如下溶液,所述溶液含有與米托蒽醌二鹽酸鹽相同的溶劑中的0.03毫摩爾%的na+(r1so3)-或na+(r2oso3)-。目測監測黑色膠體的形成。膠體緩慢分解成黑色沉淀物和灰白色上清液。在完成添加之后,獲得的混合物攪拌一段額外時間(1-7天)。膠體組合物直接使用或者存儲在冰箱中以待后用。通過uv法,以662、611或242nm確定膠體中米托蒽醌的濃度。對于取樣,用甲醇稀釋膠體的等分試樣至>20:1。實施例3:制備膠體伊立替康硫酸烷基酯和磺酸鏈烷酯在室溫下,向去離子水中的伊立替康鹽酸鹽三水合物的劇烈溶液(5ml,4mg/ml)中添加含有去離子水中的na+(r1so3)-或na+(r2oso3)-的溶液。目測監測膠體的形成。在完成添加之后,獲得的混合物攪拌2天,以及混合物以3000rpm離心10分鐘。靜置時,白色膠體緩慢分解成白色沉淀物和近乎無色的上清液。用5%的水性右旋糖替代上清液。通過渦旋10分鐘,使得沉淀物重懸浮在水中。然后,獲得的組合物直接使用或者存儲在冰箱中以待后用。通過uv法,以360、255或220nm確定膠體或改性膠體中伊立替康的濃度。對于取樣,用甲醇稀釋產物的等分試樣(甲醇過量>20:1)。實施例4:制備膠體長春瑞濱硫酸烷基酯和磺酸鏈烷酯在室溫下,向錐形燒瓶中的5%水性右旋糖中的長春瑞濱酒石酸鹽的劇烈攪拌溶液(2ml,5mg/ml)添加與長春瑞濱酒石酸鹽相同的溶劑中的一當量的na+(r1so3)-或na+(r2oso3)-的溶液。目測監測膠體的形成。在完成添加之后,獲得的混合物渦旋或振蕩7天。靜置時,膠體緩慢分解成沉淀物和透明上清液。膠體直接使用或者存儲在冰箱中以待后用。通過uv法,以268或212nm確定膠體中長春瑞濱的濃度。對于取樣,用甲醇稀釋膠體的等分試樣(甲醇過量>20:1)。實施例5:膠體阿霉素硫酸烷基酯和磺酸鏈烷酯在30%水性乙醇中的溶解度根據材料和方法中所述的一般方法確定溶解度。結果總結見表1并如圖1和2所示。表1:膠體阿霉素硫酸烷基酯和磺酸鏈烷酯在30%乙醇中的溶解度(v/v)實施例6:膠體米托蒽醌硫酸烷基酯和磺酸鏈烷酯在30%水性乙醇中的溶解度根據材料和方法中所述的一般方法確定溶解度。結果總結見表4并如圖3和4所示。表2:膠體米托蒽醌硫酸烷基酯和磺酸鏈烷酯在30%乙醇中的溶解度(v/v)實施例7:伊立替康磺酸鏈烷酯在10%水性乙醇中的溶解度根據材料和方法中所述的一般方法確定溶解度。結果總結見表3和圖5。表3:膠體伊立替康磺酸鏈烷酯在30%乙醇中的溶解度(v/v)烷基鏈長度,n溶解度,mg/ml100.83483120.15453140.07064160.01476實施例8:膠體長春瑞濱磺酸鏈烷酯在20%水性乙醇中的溶解度根據材料和方法中所述的一般方法確定溶解度。結果總結見表4和圖6。表4:膠體長春瑞濱磺酸鏈烷酯在20%水性乙醇中的溶解度烷基鏈長度,n溶解度,mg/ml101.11868120.25692140.06731160.01977實施例9:阿霉素在wistar大鼠肝臟的分布的體內分析將阿霉素鹽酸鹽的水性溶液、阿霉素硫酸鏈烷酯的水性膠體、以及阿霉素磺酸烷基酯的水性膠體經由單劑注射給藥到尾部中。動物在經由單劑注射給藥到尾部中之后2小時死亡;總阿霉素劑量為5mg/kg。根據上文所述過程確定肝臟中的阿霉素濃度。結果總結見表5和圖7。表5:阿霉素在wistar大鼠肝臟中的分布的體內分析實施例10:阿霉素在加州兔肝臟中的分布的體內分析以1ml/分鐘的緩慢流動速率將阿霉素和阿霉素鹽酸鹽的膠體非共價絡合物給藥到加洲兔的邊緣耳靜脈,阿霉素的總劑量是5mg/kg。動物在給藥后0.5小時死亡。根據上文所述過程確定肝臟中的阿霉素濃度。結果參見表6和圖8。表6:加洲兔肝臟中的阿霉素分布實施例11:制備包含在30%水性乙醇中具有所需溶解度的阿霉素磺酸鹽/酯的膠體該實施例闡述了在特定溶劑中制備具有所需溶解度的本發明的膠體。設定如下參數:活性劑:阿霉素;陰離子:具有偶數數量的碳原子的磺酸鏈烷酯;溶劑:30%水性乙醇;所需溶解度:0.1mg/ml。基于中等鏈長的磺酸鏈烷酯基團中的碳原子數量是相加的假設進行計算。還基于30%水性乙醇中的溶解度y的連續函數的假設來進行計算。根據由實施例5(參見圖2)獲得的函數(f1),表示了碳原子數量x與溶解度y之間的關系。y=f1(x)=1754.71710exp(-0.74429x)(方程式1)函數(f2)進行逆計算,即從給定溶解度y確定碳原子數量x:x=f2(y)=-1/0.7442855697ln(y/1754.71709855)(方程式2)對于y=0.1mg/ml的溶解度,函數f2得出x等于13.20628。基于基團中碳原子數量的相加效應的假定,c12和c14磺酸鹽/酯的比例行為(磺酸鹽/酯對具有最接近所需情況的溶解度),提供了所示的c13.20628基團:一當量的c13.20628等于0.397當量的c12和0.603當量的c14的混合物。根據實施例1所述的一般方法來制備具有確定的c12和c14比例的阿霉素磺酸鹽/酯的混合物。根據上文所述的一般方法確定溶解度,溶解度是0.098713mg/ml。實施例12:本發明的組合物靜脈靶向遞送到加洲兔的肝臟通過注入耳靜脈,將根據實施例1的本發明的阿霉素組合物(r=c16磺酸鹽/酯)給藥到加洲兔,濃度為1.25mg阿霉素/kg體重。作為對比,以相同方式給藥水性阿霉素鹽酸鹽,濃度為5.0mg阿霉素/kg體重。如圖9所示,對于將阿霉素遞送到肝臟,本發明的組合物比現有技術阿霉素鹽酸鹽更為有效4倍,而對于遞送到心臟而言沒有差別。當前第1頁12
相關技術
網友詢問留言
已有0條留言
- 還沒有人留言評論。精彩留言會獲得點贊!
1
- 含叔胺藥物物質的靶向遞送的制造方法與工藝
- 一種磁靶向雙載藥遞釋系統及其制備方法與流程
- 用于引導和靶向按需物質遞送的納米結構的載體的制造方法與工藝
- 附載蟾毒靈的多肽修飾的聚甲基丙烯酸寡聚乙二醇酯-聚己內酯納米制劑的制作方法
- 附載蟾毒靈的多肽修飾的聚甲基丙烯酸寡聚乙二醇酯-聚己內酯納米制劑的制作方法
- 一種用于治療去勢抵抗性前列腺癌的靶向前藥及其納米制劑和制備方法
- 一種具有內涵體逃逸功能的腫瘤靶向前藥及其納米制劑和制備方法
- 具有腦腫瘤靶向和腫瘤組織穿透能力的d構型多肽及其基因遞釋系統的制作方法
- 聚乙二醇-聚乳酸羥基乙酸-聚賴氨酸納米遞送系統、制備方法及其應用的制作方法
- 載蟾蜍靈的環肽修飾的聚乙二醇-聚乳酸羥基乙醇酸-聚賴氨酸納米粒的制作方法
- 靶向PD-1的多肽及其應用的制造方法與工藝
- 靶向同位素生產系統的制造方法與工藝
- 在制備68GA?螯合物官能化的靶向劑中用作金屬抑制劑的單糖、二糖或多糖的制造方法與工藝
- 用于引導和靶向按需物質遞送的納米結構的載體的制造方法與工藝
- 通過PROSTRATIN靶向K?RAS介導的信號轉導通路和惡性腫瘤的制造方法與工藝
- 腫瘤靶向核磁共振/熒光雙模態成像造影劑及其制備和應用的制造方法與工藝
- pH氧化還原雙敏感的PAMAM靶向納米給藥載體及其制備方法與制造工藝
- 具有卵巢腫瘤靶向D構型多肽及其基因遞釋系統的制造方法與工藝
- 靶向survivin納米抗體及其制備方法和應用與制造工藝
- 用于PSMA靶向的成像和放射療法的金屬/放射性金屬標記的PSMA抑制物的制造方法與工藝