六硼化鑭納米粉體的制備方法及應用與流程
本發明屬于陶瓷粉體制備技術領域,尤其涉及一種六硼化鑭納米粉體的制備方法及應用。
背景技術:
以六硼化鑭(LaB6)為代表的稀土金屬六硼化物具有熔點高、硬度大、逸出功低、蒸發速率低、化學性能穩定及耐離子轟擊等優點,是極佳的陰極電子發射體材料,在電子束焊機、等離子體源、掃描電鏡及俄歇譜儀等儀器中具有廣闊的應用前景。目前,常用的LaB6陰極電子發射體材料主要是由LaB6粉體經高溫燒結而成的,但現有的LaB6粉體存在粒徑粗大、尺寸不均、易團聚及雜質多等缺點,這些缺點一方面導致其燒結難度大,難以致密化,另一方面雜質的存在,特別是氧和碳,會導致LaB6陰極電子發射體材料的電子逸出功增大,從而使其電子發射性能降低。因此,為獲得高致密、高性能的LaB6陰極電子發射體材料,制備出具有高比表面積、粒徑均勻、分散性好及高純度等特性的LaB6納米粉體是關鍵。
目前常見的LaB6粉體的制備方法有:純元素化學合成法(張粹偉. 六硼化鑭陰極. 光電子技術,1989,9(3):35-44.)、硼/碳熱還原法(鄭樹起,閔光輝,鄒增大,等. 硼熱還原法制備LaB6粉末. 硅酸鹽學報,2001,29(2):128-131.)、自蔓延高溫合成法(張廷安,豆志河,楊歡. 自蔓延高溫合成LaB6微粉的制備及表征. 東北大學學報,2005,26(1):67-69.)及硼氫化鈉熱還原法(Y.F. Yuan, L. Zhang, L.M. Liang, et al. A solid-state reaction route to prepare LaB6 nanocrystals in vacuum. Ceramics International, 2011, 37:2891-2896.)等。這些方法都屬于固相反應法范疇,而固相反應法存在反應溫度高,原子擴散路徑長,反應不完全等缺點,易導致制備的粉體粒徑大,顆粒不均勻,易團聚,且雜質多,表面活性低等。除上述方法外,專利CN101503198 B公開了一種固相反應低溫合成六硼化鑭納米粉體的方法,該方法制備溫度低,能夠獲得納米尺寸的LaB6粉體,但其所需反應物種類多,易造成合成的LaB6粉體所含雜質多。
技術實現要素:
本發明要解決的技術問題是克服現有技術的不足,提供一種納米尺寸、粒徑均勻、無團聚、純度高的六硼化鑭納米粉體的制備方法,還提供該制備方法所制備的六硼化鑭納米粉體在制備陰極電子發射體中的應用。
為解決上述技術問題,本發明采用以下技術方案:
一種六硼化鑭納米粉體的制備方法,包括以下步驟:
將鑭源、硼源和熔鹽混合后加熱至熔融狀態,使鑭源和硼源在熔融鹽中發生液相反應,鑭源被硼源還原為六硼化鑭,冷卻后得到初產品;將所述初產品進行洗滌和過濾,以去除熔鹽,干燥后得到六硼化鑭納米粉體。
上述的六硼化鑭納米粉體的制備方法,優選的,所述鑭源為含鑭無機鹽。
上述的六硼化鑭納米粉體的制備方法,優選的,所述含鑭無機鹽包括LaCl3或La(NO3)3。
上述的六硼化鑭納米粉體的制備方法,優選的,所述硼源為堿金屬硼氫化物。
上述的六硼化鑭納米粉體的制備方法,優選的,所述堿金屬硼氫化物包括LiBH4、NaBH4或KBH4。
上述的六硼化鑭納米粉體的制備方法,優選的,所述鑭源中的鑭與所述硼源中的硼的摩爾比為1∶6~12。
上述的六硼化鑭納米粉體的制備方法,優選的,所述熱處理的具體過程為:在真空或惰性氣體氣氛下,以3℃/min~20℃/min的升溫速率升溫至500℃~1200℃,保溫1h~5h,再以2℃/min~15℃/min的降溫速率降至室溫。
上述的六硼化鑭納米粉體的制備方法,優選的,所述熔鹽為LiCl、KCl和NaCl中的一種或兩種。
上述的六硼化鑭納米粉體的制備方法,優選的,所述熔鹽的質量為鑭源和硼源總質量的3~15倍。
上述的六硼化鑭納米粉體的制備方法,進一步地,所述洗滌的條件為:洗滌液為去離子水或蒸餾水,洗滌溫度為20℃~90℃。
上述的六硼化鑭納米粉體的制備方法,進一步地,熔鹽去除的標準為:將AgNO3溶液滴入過濾后的洗滌液中,無白色沉淀產生。
作為一個總的發明構思,本發明還提供一種上述的六硼化鑭納米粉體的制備方法所制備的六硼化鑭納米粉體在制備陰極電子發射體中的應用。
所述應用包括作為制備等離子源、電子束焊機、電子束曝光機及各類電鏡等設備的陰極元器件的原材料,還包括作為等離子源、電子束焊機、電子束曝光機及各類電鏡等設備的陰極元器件的涂層材料。
與現有技術相比,本發明的優點在于:
1、本發明的六硼化鑭納米粉體的制備方法,通過熔鹽法提供的高溫熔融鹽液相環境,一方面使鑭源與硼源反應更快、更加充分,另一方面實現對鑭源與硼源反應生成LaB6的形核與生長速度的控制,從而制備出純度高、粒徑細小均一、分散性好等的LaB6納米粉體,巧妙地避開了現有固相反應法制備LaB6粉體存在的反應溫度高,原子擴散路徑長,反應不完全等固有難題。
2、本發明的六硼化鑭納米粉體的制備方法,鑭金屬源優選為LaCl3或La(NO3)3,硼源優選為堿金屬硼氫化物,熔鹽介質優選為堿金屬氯化物。一方面,從原料體系和熔鹽體系都較好地規避了氧和碳的攝入,使得制備出的LaB6納米粉體的純度高,而以碳熱還原法制備六硼化鑭粉體(La2O3 + 6 B2O3 + 21 C = 2 LaB6 + 21 CO)為例,其原材料中帶有的氧和碳會因為反應不完全或實際原料配比與理論配比存在差異而殘留在所制備的LaB6粉體中,且很難去除。另一方面,本發明優選的鑭金屬源和硼源在熔鹽中都具有較大的溶解度,兩者反應生成LaB6的機制為溶解-析出機制,即鑭源和硼源在液態熔融鹽中以離子態的形式混合并反應生成LaB6,而生成的LaB6經形核、生長,最終從熔鹽中析出,因此通過調整工藝條件控制LaB6的形核和生長速度,即可實現LaB6納米粉體的可控制備,與一般的模板合成機制(即反應原材料中有一種或多種在熔鹽中的溶解度小,導致原材料之間的反應是通過以溶解度小的原材料為核,其他原材料擴散到其表面并反應生成目標產物的,隨著反應的進行,目標產物逐漸取代原反應原材料核)相比,對合成原料的粒徑要求不高,所形成的晶核為納米級,且形核均勻。
3、本發明的六硼化鑭納米粉體的制備方法,由于反應產物LaB6的形核和生長是在液態的熔融鹽中進行的,除原材料在熔鹽中的溶解度、反應機制等影響外,高溫熔融鹽液相環境條件(溫度、時間及降溫速率等)也會對反應產物LaB6的形核和生長有極大的影響。試驗表明,液態熔融鹽介質的溫度越高或保溫時間越長,制備的LaB6粉體的粒徑越大。除此之外,降溫速率也對其影響很大,降溫速率過快,得到的粉體細小,但可能結晶度低;降溫速率過慢,則易導致粉體粗大且團聚。因此,本發明通過嚴格控制好高溫熔融鹽的溫度(500℃~1200℃)、保溫時間(1h~5h)及降溫速率(2℃/min~15℃/min)等,可以制備得到一系列不同粒徑的LaB6納米粉體。
4、本發明的六硼化鑭納米粉體的制備方法,所制備的LaB6納米粉體粒徑細小均一,粒徑范圍在10nm~200nm之間,且純度高,分散性好。此粉體可作為熱壓燒結或放電等離子燒結制備致密LaB6塊體材料的優選原料,也可以用于制備LaB6涂層及其他復合材料等領域。尤其在制備等離子源、電子束焊機、電子束曝光機及各類電鏡等設備的陰極元器件用六硼化鑭塊體或涂層陰極電子發射體方面有明顯的優勢。
5、本發明的六硼化鑭納米粉體的制備方法,原材料來源廣,反應溫度低,工藝過程簡單可控,無需特殊設備要求,成本低等優勢,適合工業化生產。
附圖說明
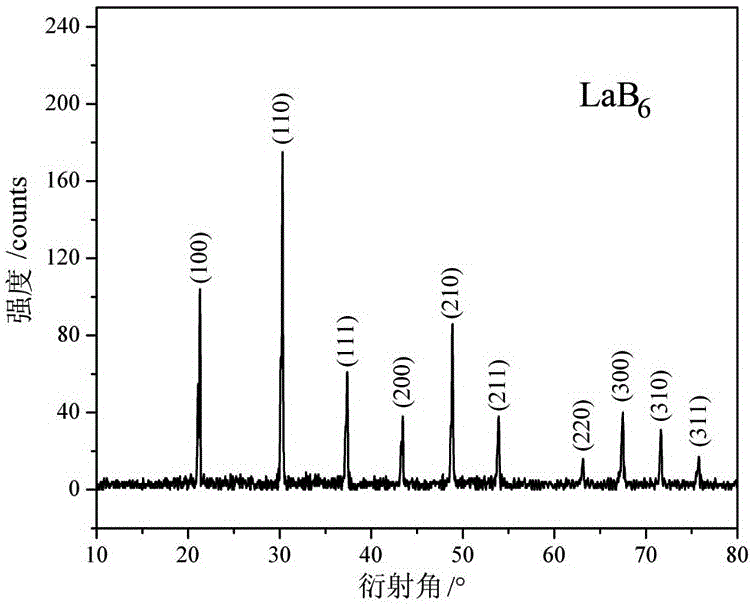
圖1為本發明實施例1制備的LaB6納米粉體的XRD譜圖。
圖2為本發明實施例1制備的LaB6納米粉體的SEM照片。
圖3為本發明實施例1制備的LaB6納米粉體的粒徑分布圖。
具體實施方式
以下結合說明書附圖和具體優選的實施例對本發明作進一步描述,但并不因此而限制本發明的保護范圍。
實施例1:
一種本發明的六硼化鑭納米粉體的制備方法,包括以下步驟:
(1)按La∶B摩爾比為1∶6的比例,分別稱取1.23g LaCl3,1.14g NaBH4,按KCl-LiCl復相鹽的總質量為LaCl3和NaBH4的總質量的5倍,分別稱取6.64g KCl,5.21g LiCl,將四者充分研磨混合均勻后裝入坩堝;
(2)將裝料后的坩堝放置于管式爐中進行熱處理,在氬氣保護氣氛下,以5℃/min的升溫速率升溫至600℃,保溫1h,之后以8℃/min的速率降溫至室溫,得到初產物;
(3)采用60℃的去離子水對初產物進行多次洗滌,直至將AgNO3溶液滴加至過濾后的洗滌液中時無白色沉淀為止,最后進行干燥,得到六硼化鑭納米粉體。
本實施例所制備的六硼化鑭納米粉體經X射線衍射分析為純LaB6相,不含其它雜質相,如圖1所示。該六硼化鑭納米粉體的掃描電鏡照片如圖2所示,由圖可知,粉體顆粒均勻、無團聚,粒徑范圍為10nm~40nm。通過對粉體的粒徑分布做統計,結果如圖3所示,粉體顆粒均勻,大部分的粉體粒徑集中在10nm~25nm之間。
實施例2:
一種本發明的六硼化鑭納米粉體的制備方法,包括以下步驟:
(1)按La∶B摩爾比為1∶8的比例,分別稱取1.63g La(NO3)3,1.52g NaBH4,按KCl-LiCl復相鹽的總質量為La(NO3)3和NaBH4的總質量的10倍,分別稱取17.33g KCl,14.17g LiCl,將四者充分研磨混合均勻后裝入坩堝;
(2)將裝料后的坩堝放置于管式爐中進行熱處理,在氬氣保護氣氛下,以5℃/min的升溫速率升溫至800℃,保溫1h,之后以5℃/min的速率降溫至室溫,得到初產物;
(3)采用60℃的去離子水對初產物進行多次洗滌,直至將AgNO3溶液滴加至過濾后的洗滌液中時無白色沉淀為止,最后進行干燥,得到六硼化鑭納米粉體。
經檢測分析,本實施例所制備的六硼化鑭納米粉體為純LaB6相,粒徑范圍為80nm~120nm。
實施例3:
一種本發明的六硼化鑭納米粉體的制備方法,包括以下步驟:
(1)按La∶B摩爾比為1∶6的比例,分別稱取1.23g LaCl3,1.14g NaBH4,按KCl單相鹽的質量為LaCl3與NaBH4的總質量的5倍,稱取11.85g KCl,將三者充分研磨混合均勻后裝入坩堝;
(2)將裝料后的坩堝放置于管式爐中進行熱處理,在氬氣保護氣氛下, 以5℃/min的升溫速率升溫至1000℃,保溫2h,之后以5℃/min的速率降溫至室溫,得到初產物;
(3)采用60℃的去離子水對初產物進行多次洗滌,直至將AgNO3溶液滴加至過濾后的洗滌液中時無白色沉淀為止,最后進行干燥,得到六硼化鑭納米粉體。
經檢測分析,本實施例所制備的六硼化鑭納米粉體為純LaB6相,粒徑范圍為140nm~200nm。
實施例4:
一種本發明的六硼化鑭納米粉體的制備方法,包括以下步驟:
(1)按La∶B摩爾比為1∶6的比例,分別稱取1.23g LaCl3,1.14g NaBH4,按KCl-LiCl復相鹽的總質量為LaCl3和NaBH4的總質量的5倍,分別稱取6.64g KCl,5.21g LiCl,將四者充分研磨混合均勻后裝入坩堝;
(2)將裝料后的坩堝放置于管式爐中進行熱處理,在氬氣保護氣氛下,以5℃/min的升溫速率升溫至600℃,保溫1h,之后以2℃/min的速率降溫至室溫,得到初產物;
(3)采用60℃的去離子水對初產物進行多次洗滌,直至將AgNO3溶液滴加至過濾后的洗滌液中時無白色沉淀為止,最后進行干燥,得到六硼化鑭納米粉體。
經檢測分析,本實施例所制備的六硼化鑭納米粉體為純LaB6相,粒徑范圍為40nm~70nm。
以上所述僅是本發明的優選實施方式,本發明的保護范圍并不僅局限于上述實施例。凡屬于本發明思路下的技術方案均屬于本發明的保護范圍。應該指出,對于本技術領域的普通技術人員來說,在不脫離本發明原理的前提下的改進和潤飾,這些改進和潤飾也應視為本發明的保護范圍。
- 還沒有人留言評論。精彩留言會獲得點贊!