一種改善鉍鐵鈦類氧化物納米粉體團聚的方法與流程
本發明涉及無機材料技術領域,特別涉及一種改善鉍鐵鈦類氧化物納米粉體團聚的方法。
背景技術:
鉍鐵鈦類氧化物是一類單相的具有層狀鈣鈦礦晶體結構的化合物,通式為bim+1fem-3ti3o3m+3(m≤10),其同時具有極化有序、磁化有序及寬光譜響應等多種性能。另外,該類氧化物還作為一類新興的多功能光催化劑而被廣泛關注,其相比于傳統光催化劑具有如下獨特優勢:①具有良好的太陽光光譜吸收性;②內部的自發極化有助于光生載流子的分離而使光催化性能提高;③其室溫磁性使其能夠從反應液中分離后循環使用,有利于節約資源,避免二次污染。2008年,王文中等報道了bi5feti3o15的可見光光催化降解有機染料;陸亞林等報道了co摻雜的bi7fe3ti3o21可見-近紅外輻射下的羅丹明b染料光降解,且實現了從高溫粘度溶液中的磁性分離和回收。然而,進一步提高鉍鐵鈦類氧化物的光催化活性仍是當前研究的重點。
光催化活性不僅取決于材料的結構形貌,也受到納米顆粒分散性的影響。由于納米顆粒的小尺寸效應等因素,顆粒表面活性高而易團聚,導致有效比表面積降低、反應活性位點減少;另外,鉍鐵鈦類氧化物具有鐵磁有序性,在液相法(如水熱法、共沉淀法、溶膠凝膠法即微乳液法等)從下而上的晶粒生長過程中,晶粒之間存在磁性吸引力,會進一步加劇納米小尺寸效應引起的團聚現象。眾所周知,粉體的團聚嚴重制約著材料的表觀性能,尤其是對于以固-液反應體系為主的光催化反應。因此,降低甚至抑制粉體顆粒團聚,對于提高光催化性能具有重要意義。
現有技術中,在改善鉍鐵鈦類氧化物粉體的團聚問題時通常是在制備產物過程中通過改變制備條件如改變前驅體濃度、反應溫度及時間、ph值、表面活性劑、洗滌干燥方式等來改善,其中,最主要的是在液相合成過程中加入有機表面活性劑或阻擋劑,這些表面活性劑選擇性的吸附在晶粒表面,由于空間位阻效應,擴大了晶粒之間的距離,理論上可以在合成時控制團聚現象;但據目前文獻報道,在實際應用時,即使使用表面活性劑也難以抑制粉體顆粒的團聚;另外,就算在合成時團聚現象有一定改善,隨后將氧化物粉體顆粒從液相中分離收集的過程中,由于納米小尺寸效應等復雜誘因,還會再次出現團聚現象。因此,對于鉍鐵鈦類氧化物納米粉體,在合成過程及顆粒收集過程中,均難以通過調控實驗條件來改善團聚問題。
技術實現要素:
有鑒于此,本發明的目的在于提供一種改善鉍鐵鈦類氧化物納米粉體團聚的方法,采用本發明的方法能夠有效改善鉍鐵鈦類氧化物粉體中納米顆粒的團聚問題,獲得單分散性良好的鉍鐵鈦類氧化物納米顆粒。
本發明提供了一種改善鉍鐵鈦類氧化物納米粉體團聚的方法,包括以下步驟:
a)將鉍鐵鈦類氧化物納米粉體與酸液混合,得到懸浮液;
b)將所述懸浮液密封靜置或密封攪拌,得到混合液;
c)將所述混合液超聲分散,得到分散液;
d)將所述分散液離心、洗滌和冷凍干燥,得到分散產物。
優選的,所述步驟a)中,所述酸液為乙酸溶液或鹽酸溶液。
優選的,所述步驟a)中,所述酸液的濃度為0.1%~3%。
優選的,所述步驟a)中,鉍鐵鈦類氧化物納米粉體的質量與酸液的體積比為0.1g:(20~50)ml。
優選的,所述步驟b)中,所述密封靜置或密封攪拌的時間為1~6h。
優選的,所述步驟b)中,所述密封攪拌的速率為100~300r/min。
優選的,所述步驟c)中,超聲分散的功率為40~80w,時間為1~2h。
優選的,所述步驟c)中,超聲分散的溫度為30℃以下。
優選的,所述步驟c)中,超聲分散的頻率為40~60khz。
優選的,所述步驟d)中,離心的速率為7000~12000r/min。
本發明提供了一種改善鉍鐵鈦類氧化物納米粉體團聚的方法,包括:a)將鉍鐵鈦類氧化物納米粉體與酸液混合,得到懸浮液;b)將所述懸浮液密封靜置或密封攪拌,得到混合液;c)將所述混合液超聲分散,得到分散液;d)將所述分散液離心、洗滌和冷凍干燥,得到分散產物。采用本發明的方法能夠有效改善鉍鐵鈦類氧化物粉體的團聚問題,并且不會破壞鉍鐵鈦類氧化物顆粒的晶體結構和形貌,能夠獲得單分散性良好的鉍鐵鈦類氧化物納米顆粒。
附圖說明
為了更清楚地說明本發明實施例或現有技術中的技術方案,下面將對實施例或現有技術描述中所需要使用的附圖作簡單地介紹,顯而易見地,下面描述中的附圖僅僅是本發明的實施例,對于本領域普通技術人員來講,在不付出創造性勞動的前提下,還可以根據提供的附圖獲得其他的附圖。
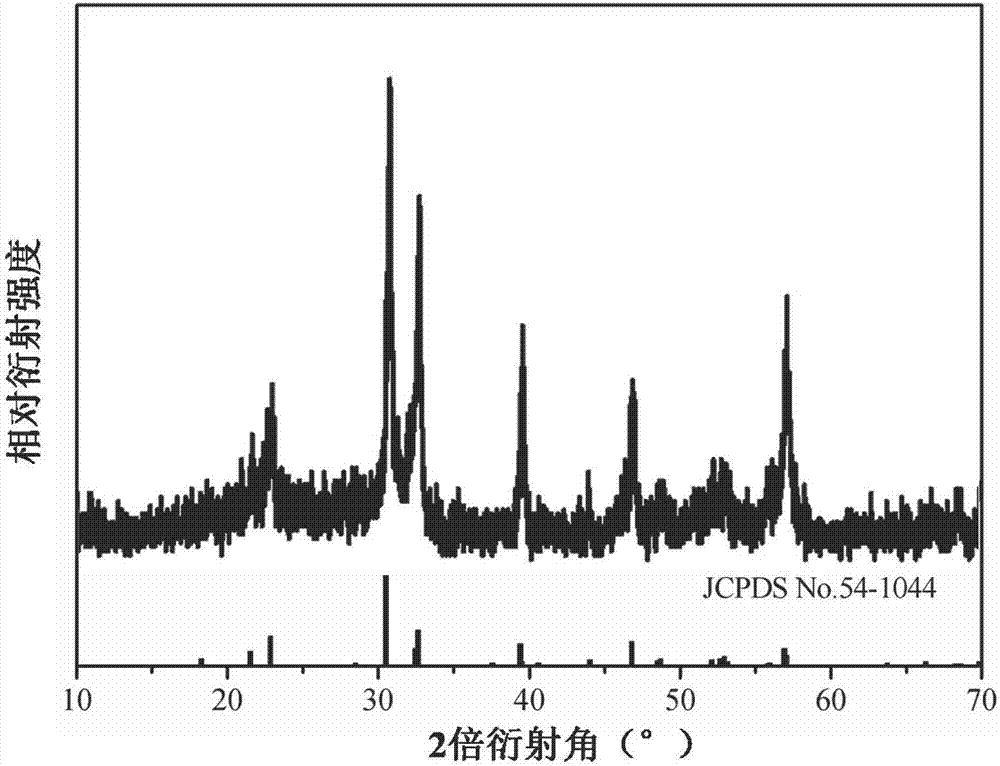
圖1為本發明實施例1中樣品d的x射線衍射圖;
圖2為本發明實施例1中樣品s的x射線衍射圖;
圖3為本發明實施例1中樣品d的sem測試圖;
圖4為本發明實施例1中樣品s的sem測試圖;
圖5為本發明實施例1中分散處理前、后樣品的掃描電子顯微對比圖。
具體實施方式
本發明提供了一種改善鉍鐵鈦類氧化物納米粉體團聚的方法,包括以下步驟:
a)將鉍鐵鈦類氧化物納米粉體與酸液混合,得到懸浮液;
b)將所述懸浮液密封靜置或密封攪拌,得到混合液;
c)將所述混合液超聲分散,得到分散液;
d)將所述分散液離心、洗滌和冷凍干燥,得到分散產物。
采用本發明的方法能夠有效改善鉍鐵鈦類氧化物粉體的團聚問題,并且不會破壞鉍鐵鈦類氧化物顆粒的晶體結構和形貌,能夠獲得單分散性良好的鉍鐵鈦類氧化物納米顆粒。
按照本發明,首先將鉍鐵鈦類氧化物納米粉體與酸液混合,得到懸浮液。
本發明中,所述鉍鐵鈦類氧化物的種類沒有特殊限制,為本領域中的常規鉍鐵鈦類氧化物即可,即滿足通式bim+1fem-3ti3o3m+3(m≤10)即可,包括但不限于bi7fe3ti3o21、bi5feti3o15等。
本發明中,所述鉍鐵鈦類氧化物納米粉體的獲得方式沒有特殊限制,為由本領域中的常規制備方法制得的納米粉體即可,如可以為由微乳液法、共沉淀法、水熱法或溶膠凝膠法制得的鉍鐵鈦類氧化物納米粉體。
以bi7fe3ti3o21納米粉體為例,采用共沉淀法制備時,可參考文獻“liu,z.;qi,y.;lu,c.,highefficientultravioletphotocatalyticactivityofbifeo3nanoparticlessynthesizedbyachemicalcoprecipitationprocess.journalofmaterialsscience:materialsinelectronics2009,21,380-384”公開的共沉淀法制備鉍鐵氧化物的制備過程,對原料進行適應性調整即可;例如,具體的可按照如下過程制備:將鉍源化合物(如硝酸鉍)、鐵源化合物(硝酸鐵)和鈦源化合物(如鈦酸四丁酯)按照原子摩爾比例bi:fe:ti=7:3:3的比例混合,共同溶解于濃度為4m的硝酸液中形成混合溶液,再逐滴加入氫氧化鈉溶液(濃度1m),反應形成懸浮液;將懸浮液陳化24小時后去除上清液,用去離子水和無水乙醇反復洗滌至ph約為7時,在60℃下干燥,700℃下煅燒2小時,隨爐冷卻;之后分散于乙醇中超聲分散2小時,取均勻混合液干燥蒸發溶劑后得到bi7fe3ti3o21納米粉體。采用溶膠凝膠法制備bi7fe3ti3o21納米粉體時,可參考文獻“sun,s.j.;wang,g.p.;huang,y.;wang,j.l.;peng,r.r.;lu,y.l.,structuraltransformationandmultiferroicpropertiesingd-dopedbi7fe3ti3o21ceramics.rscadv.2014,4,30440-30446”公開的方法進行。采用水熱法制備bi7fe3ti3o21納米粉體時,可參考文獻“li,x.;ju,z.;li,f.;huang,y.;xie,y.;fu,z.;knized,r.j.;lu,y.,visiblelightresponsivebi7fe3ti3o21nanoshelfphotocatalystswithferroelectricityandferromagnetism.j.mater.chem.a2014,2,13366-13372.”公開的方法進行。以bi5feti3o15納米粉體為例,采用共沉淀法制備時,可參考上述bi7fe3ti3o21納米粉體的共沉淀方法進行,根據目標產物中各原子比例對各原料化合物的添加量進行相應調整即可。
本發明中,所用酸液優選為乙酸溶液或鹽酸溶液;采用所述兩種酸液有利于破壞鉍鐵鈦類氧化物納米粉體團聚顆粒界面的弱化學鍵,促進顆粒分離。
本發明中,所述酸溶液的濃度優選為0.1%~3%,更優選為0.39%~1.92%;濃度過低不能有效破壞團聚顆粒界面處的鍵合,濃度過高,h+易侵蝕鉍鐵鈦類氧化物納米顆粒的晶體結構,材料的物相將發生變化,甚至氧化物顆粒全部溶解于酸性溶液中。本發明中所述酸液可以為由市售酸液與水配制而得,例如,可將市售高濃度酸液(如質量濃度為98%的乙酸或為36%~38%的鹽酸)與水按體積比為(4~20):1000獲得,或者由低濃度酸液與水按相應體積比配制獲得,對此沒有特殊限制。
經發明人長期研究發現,本發明采用特定種類及濃度的酸液處理一定量的鉍鐵鈦類氧化物納米粉體,能夠使團聚顆粒之間的橋氧鍵斷裂,破壞團聚顆粒界面間的弱化學鍵,及減弱納米顆粒的小尺寸效應,且不會破壞納米顆粒的晶體結構和形貌,有利于獲得單分散性良好的納米粉體。
本發明中,將鉍鐵鈦類氧化物納米粉體與酸液混合時,鉍鐵鈦類氧化物納米粉體的質量與酸液的體積比優選為0.1g:(20~50)ml。本發明中,所述混合的方式沒有特殊限制,按照常規粉液混合方式進行即可,如可以邊攪拌酸液邊緩慢加入鉍鐵鈦類氧化物納米粉體,使粉體在酸液中分布均勻,進而獲得懸浮液。
按照本發明,在得到懸浮液后,將所述懸浮液密封靜置或密封攪拌,得到混合液。
本發明中,密封的方式沒有特殊限制,按照本領域中的常規密封方式進行即可,如可以在懸浮液容器開口處用密封膜進行密封等。進行密封,能夠防止酸揮發,還能使密閉空間內被酸性氣體填充,有利于增大酸性氣體蒸汽壓,進而有利于酸在溶液中電離形成h+,作用于團聚顆粒之間。
本發明中,密封靜置的時間優選為1~6小時。本發明中,優選進行密封攪拌,密封攪拌時,優選為低速磁力攪拌;所述攪拌的轉速優選為100~300r/min。本發明中,所述密封攪拌的時間優選為1~6小時。通過密封時間的控制,有利于保障h+對顆粒團聚界面的化學鍵進行有效及適度的反應和作用,進而有利于提高最終分散效果。
按照本發明,得到混合液后,將所述混合液超聲分散,得到分散液。
本發明中,所述超聲分散的功率優選為40~80w。所述超聲分散的時間優選為1~2小時。所述超聲分散的頻率優選為40~60khz。本發明中,所述超聲分散優選在30℃以下進行。通過上述超聲分散,獲得分布均勻的分散液。
按照本發明,得到分散液后,將所述分散液離心、洗滌和冷凍干燥,得到分散產物。
本發明中,所述離心優選為高速離心,具體優選為在7000~12000r/min的高速下離心。本發明中,在離心后,對離心所得固體物質進行洗滌;所述洗滌優選采用去離子水洗滌;所述洗滌的次數沒有特殊限制,優選洗滌至洗滌液的ph值約為7,此時去除洗滌液或上清液,得到固體洗滌物。
本發明中,在洗滌后,優選將固體洗滌物與水混合,形成固液混合物;本發明中,所述固體洗滌物的質量與水的體積比優選為0.1g:(20~50)ml。所述水優選為去離子水。所述混合的方式沒有特殊限制,能夠使固液混合均勻即可。
本發明中,在得到固液混合物后,再進行冷凍干燥;本發明中,所述冷凍干燥的方式及條件沒有特殊限制,按照常規的冷凍干燥方式進行即可。本發明中,待冷凍干燥、冰塊升華后,即得到單分散性良好的鉍鐵鈦類氧化物納米粉體。
實驗結果表明,按照本發明的方法處理原始鉍鐵鈦類氧化物納米粉體,能夠明顯改善納米顆粒的團聚現象,原始團聚至微米尺度的粉體經處理后,粉體中納米尺寸的晶粒彼此獨立存在,具有高度單分散性。而且,經處理后,粉體的x射線圖譜仍與標準圖譜一致,物相較純,證明本發明的分散處理并未改變或破壞bi7fe3ti3o21納米顆粒的晶體結構。
為了進一步理解本發明,下面結合實施例對本發明優選實施方案進行描述,但是應當理解,這些描述只是為進一步說明本發明的特征和優點,而不是對本發明權利要求的限制。
實施例1
1.1樣品的制備:
提供質量濃度為0.39%的乙酸溶液50ml(濃度為98%的乙酸與水按體積比4:1000配制),邊攪拌邊緩慢加入共沉淀法制得的bi7fe3ti3o21納米粉體0.1g,得到懸浮液;將所得懸浮液密封并以200r/min的轉速進行低速磁力攪拌3h,得到混合液;將所得混合液置于超聲分散器中超聲分散1小時(60w,50khz,28℃),得到分散液;將所得分散液在10000r/min的高速下離心,再用去離子水洗滌至ph約為7時去除上清液,得到固體洗滌物;將所得固體洗滌物置于50ml去離子水中混合均勻后,再冷凍干燥,待冰塊升華后,得到團聚明顯減少的單分散性良好的bi7fe3ti3o21納米粉體(記為s)。
設置對照樣:
以未進行上述分散處理的由共沉淀法制得的原始bi7fe3ti3o21納米粉體為對照樣(記為d)。
1.2樣品的表征:
(1)分別對樣品d和樣品s進行x射線衍射測試,結果分別如圖1和圖2所示(其中,圖1為樣品d的x射線衍射圖,圖2為樣品s的x射線衍射圖)。
由圖1可以看出,在實施本發明的分散處理前,原始bi7fe3ti3o21納米粉體(樣品d)的x射線圖譜與標準圖譜一致,物相較純。由圖2可以看出,在實施本發明的分散方法處理后,所得bi7fe3ti3o21納米粉體(樣品s)的x射線圖譜仍與標準圖譜一致,物相較純,證明本發明的分散處理并未改變和破壞bi7fe3ti3o21納米顆粒的晶體結構,能夠保持完整的晶體結構及形貌。
(2)分別對樣品d和樣品s進行掃描電子顯微(sem)測試,結果分別如圖3和圖4所示(其中,圖3為樣品d的sem測試圖,圖4為樣品s的sem測試圖),二者的對比效果如圖5所示(圖5為分散處理前樣品﹝即樣品d﹞、分散處理后樣品﹝即樣品s﹞的掃描電子顯微對比圖)。
由圖3可以看出,在實施本發明的分散處理前,原始bi7fe3ti3o21納米粉體(樣品d)中,顆粒團聚嚴重,數百個100nm左右的晶粒團聚達到微米尺度。由圖4可以看出,在實施本發明的分散方法處理后,所得bi7fe3ti3o21納米粉體(樣品s)中,顆粒團聚現象得到明顯改善,尺寸為100nm左右的晶粒彼此獨立存在,具有高度單分散性。由圖5也可以看出,相比于分散處理前,在實施本發明的分散方法處理后,納米粉體中顆粒團聚現象得到明顯改善。
實施例2
按照實施例1的分散方法進行,不同的是,乙酸溶液的濃度為1.92%(濃度為98%的乙酸與水按體積比20:1000配制)。
按照實施例1的表征方法對樣品進行x射線衍射測試,結果顯示,分散處理前、后,樣品的x射線圖譜均與標準圖譜一致,物相較純,證明本發明的分散處理并未改變和破壞bi7fe3ti3o21納米顆粒的晶體結構。
按照實施例1的表征方法對樣品進行掃描電子顯微(sem)測試,結果顯示,在實施本發明的分散方法處理后,所得bi7fe3ti3o21納米粉體中,顆粒團聚現象得到明顯改善,尺寸為100nm左右的晶粒彼此獨立存在,具有高度單分散性。
實施例3
按照實施例1的分散方法進行,不同的是,將乙酸溶液替換為鹽酸溶液。
按照實施例1的表征方法對樣品進行x射線衍射測試,結果顯示,分散處理前、后,樣品的x射線圖譜均與標準圖譜一致,物相較純,證明本發明的分散處理并未改變和破壞bi7fe3ti3o21納米顆粒的晶體結構。
按照實施例1的表征方法對樣品進行掃描電子顯微(sem)測試,結果顯示,在實施本發明的分散方法處理后,所得bi7fe3ti3o21納米粉體中,顆粒團聚現象得到明顯改善,尺寸為100nm左右的晶粒彼此獨立存在,具有高度單分散性。
實施例4
按照實施例1的分散方法進行,不同的是,加入的bi7fe3ti3o21納米粉體是由溶膠凝膠法制得。
按照實施例1的表征方法分別對分散處理前后的樣品進行x射線衍射測試,結果顯示,分散處理前、后,樣品的x射線圖譜均與標準圖譜一致,物相較純,證明本發明的分散處理并未改變和破壞bi7fe3ti3o21納米顆粒的晶體結構。
按照實施例1的表征方法分別對分散處理前后的樣品進行掃描電子顯微(sem)測試,結果顯示,在實施本發明的分散處理前,原始bi7fe3ti3o21納米粉體中,顆粒團聚嚴重,數百個100nm左右的晶粒團聚達到微米尺度。在實施本發明的分散方法處理后,所得bi7fe3ti3o21納米粉體中,顆粒團聚現象得到明顯改善,尺寸為100nm左右的晶粒彼此獨立存在,具有高度單分散性。
實施例5
按照實施例1的分散方法進行,不同的是,加入的是bi5feti3o15納米粉體。
按照實施例1的表征方法分別對分散處理前后的樣品進行x射線衍射測試,結果顯示,分散處理前、后,樣品的x射線圖譜均與標準圖譜一致,物相較純,證明本發明的分散處理并未改變和破壞bi5feti3o15納米顆粒的晶體結構。
按照實施例1的表征方法分別對分散處理前后的樣品進行掃描電子顯微(sem)測試,結果顯示,在實施本發明的分散處理前,原始bi5feti3o15納米粉體中,顆粒團聚嚴重,數百個100nm左右的晶粒團聚達到微米尺度。在實施本發明的分散方法處理后,所得bi5feti3o15納米粉體中,顆粒團聚現象得到明顯改善,尺寸為100nm左右的晶粒彼此獨立存在,具有高度單分散性。
實施例6
按照實施例1的分散方法進行,不同的是,將懸浮液密封并以200r/min的轉速進行低速磁力攪拌6h。
按照實施例1的表征方法對樣品進行x射線衍射測試,結果顯示,分散處理前、后,樣品的x射線圖譜均與標準圖譜一致,物相較純,證明本發明的分散處理并未改變和破壞bi7fe3ti3o21納米顆粒的晶體結構。
按照實施例1的表征方法對樣品進行掃描電子顯微(sem)測試,結果顯示,在實施本發明的分散方法處理后,所得bi7fe3ti3o21納米粉體中,顆粒團聚現象得到明顯改善,尺寸為100nm左右的晶粒彼此獨立存在,具有高度單分散性。
實施例7
按照實施例1的分散方法進行,不同的是,將混合液置于超聲分散器中超聲分散2小時。
按照實施例1的表征方法對樣品進行x射線衍射測試,結果顯示,分散處理前、后,樣品的x射線圖譜均與標準圖譜一致,物相較純,證明本發明的分散處理并未改變和破壞bi7fe3ti3o21納米顆粒的晶體結構。
按照實施例1的表征方法對樣品進行掃描電子顯微(sem)測試,結果顯示,在實施本發明的分散方法處理后,所得bi7fe3ti3o21納米粉體中,顆粒團聚現象得到明顯改善,尺寸為100nm左右的晶粒彼此獨立存在,具有高度單分散性。
實施例8
按照實施例1的分散方法進行,不同的是,乙酸溶液的濃度為0.98%(濃度為98%的乙酸與水按體積比10:1000配制);加入的bi7fe3ti3o21納米粉體是由水熱法制得。
按照實施例1的表征方法分別對分散處理前后的樣品進行x射線衍射測試,結果顯示,分散處理前、后,樣品的x射線圖譜均與標準圖譜一致,物相較純,證明本發明的分散處理并未改變和破壞bi7fe3ti3o21納米顆粒的晶體結構。
按照實施例1的表征方法分別對分散處理前后的樣品進行掃描電子顯微(sem)測試,結果顯示,在實施本發明的分散處理前,原始bi7fe3ti3o21納米粉體中,顆粒團聚嚴重,數百個100nm左右的晶粒團聚達到微米尺度。在實施本發明的分散方法處理后,所得bi7fe3ti3o21納米粉體中,顆粒團聚現象得到明顯改善,尺寸為100nm左右的晶粒彼此獨立存在,具有高度單分散性。
實施例9
按照實施例1的分散方法進行,不同的是,加入的是bi5feti3o15納米粉體;將懸浮液密封并以200r/min的轉速進行低速磁力攪拌6h;將混合液置于超聲分散器中超聲分散2小時。
按照實施例1的表征方法分別對分散處理前后的樣品進行x射線衍射測試,結果顯示,分散處理前、后,樣品的x射線圖譜均與標準圖譜一致,物相較純,證明本發明的分散處理并未改變和破壞bi5feti3o15納米顆粒的晶體結構。
按照實施例1的表征方法分別對分散處理前后的樣品進行掃描電子顯微(sem)測試,結果顯示,在實施本發明的分散處理前,原始bi5feti3o15納米粉體中,顆粒團聚嚴重,數百個100nm左右的晶粒團聚達到微米尺度。在實施本發明的分散方法處理后,所得bi5feti3o15納米粉體中,顆粒團聚現象得到明顯改善,尺寸為100nm左右的晶粒彼此獨立存在,具有高度單分散性。
由以上實施例可知,按照本發明的方法,能夠明顯改善鉍鐵鈦類氧化物(bim+1fem-3ti3o3m+3;m≤10)納米粉體的顆粒團聚現象,獲得單分散性良好的納米粉體,且不會破壞納米粉體顆粒的晶體結構。
以上實施例的說明只是用于幫助理解本發明的方法及其核心思想。對這些實施例的多種修改對本領域的專業技術人員來說將是顯而易見的,本文中所定義的一般原理可以在不脫離本發明的精神或范圍的情況下,在其它實施例中實現。因此,本發明將不會被限制于本文所示的這些實施例,而是要符合與本文所公開的原理和新穎特點相一致的最寬的范圍。
- 還沒有人留言評論。精彩留言會獲得點贊!