刺槐素的制備方法與流程
本發明涉及一種從香青蘭中提取分離得到刺槐素的方法。
背景技術:
經文獻檢索,刺槐素的提取方法已有一些文獻報道:
陳政雄(中藥黃酮類的研究VIII.野菊花成分的研究(第一報)[J].藥學學報,1962(06).)首次報道從野菊花分離得到刺槐素的結構,該工藝采用提取、萃取、重結晶、水解等技術聯合從野菊花菊中分離得到刺槐素,操作復雜、步驟繁瑣且產量低。
刺槐素的結構式為:
陳維佳(野菊花化學成分及總黃酮提取工藝研究[D].山東中醫藥大學,2009.)采用乙醇回流提取、萃取、硅膠柱、SephadexLH-20柱反復柱層析從6kg的野菊花中分離得到11.8mg刺槐素,工藝操作復雜且產量低。
丁智慧(飛機草中的化學成分[J].天然產物研究與開發,2001,13(5):22-24.)采用甲醇回流提取、萃取、硅膠柱反復柱層析從2.48kg的飛機草中分離得到0.318g刺槐素,該工藝操作步驟繁瑣。
同時,有3篇與刺槐素提取方法有關的中國專利:
王秀敏等的發明專利(從野菊花中分離制備刺槐素單體的方法:CN,CN 103193748 A[P].)采用提取、大孔樹脂柱色譜、水解、高速逆流色譜法等技術從1kg野菊花植物分離得到10mg刺槐素,該工藝采用了5種技術聯合進行分離,操作復雜、成本較高、收率低。
卞毓平等的發明專利(一種從菊花中提取刺槐素的方法:CN,CN 102491964 A[P].)采用萃取、大孔樹脂、聚酰胺柱層析和重結晶等技術從10kg菊花植物中分離得到1.2g的刺槐素,操作復雜、步驟繁瑣。
劉曉秋等的發明專利(一種柳穿魚黃酮及其總黃酮的制備方法和用途CN,CN 101475619 A[P].)采用回流提取、大孔樹脂、聚酰胺柱層析、硅膠柱層析和SephadexLH-20柱層析等技術分離得到刺槐素,該工藝采用5種技術聯合進行分離,操作復雜、步驟繁瑣。
技術實現要素:
本發明的目的是提供一種從香青蘭中提取分離得到刺槐素的方法。
本發明的目的是通過以下技術方案實現的:
a、將香青蘭干燥的地上部分粉碎,用5~10倍量的40%~95%乙醇浸泡12~24h;
b、采用滲漉法,香青蘭藥材與40%~95%乙醇比例1∶30~40,流速1~6mL/min,收集滲漉液;
c、滲漉液用孔徑0.1~10mm的網孔過濾1~5次,濾液合并;
d、用減壓蒸餾裝置,在壓力:0.08~0.1Mpa,溫度:30~60℃下,蒸餾除去乙醇得0.8~1.2g/mL的稠浸膏;水浴干燥后得浸膏粉;
e、將浸膏粉采用70%~100%乙醇溶解后干法拌樣,上100~200目聚酰胺進行柱層析,采用20~95%乙醇梯度洗脫,并收集60~75%乙醇梯度的洗脫液,每個梯度洗脫液為4~5倍量柱體積;洗脫液用減壓蒸餾裝置,在壓力:0.08~0.1Mpa,溫度:40~60℃下蒸干得浸膏粉;
f、為了進一步純化所得浸膏粉,濾去雜質,采用95%乙醇對浸膏粉進行重結晶,所得制備液用減壓蒸餾裝置,在壓力:0.08~0.1Mpa,溫度:40~60℃下蒸干得單體刺槐素。
有益技術效果
本專利采用聚酰胺柱層析和重結晶技術聯合的方法,方法簡單,步驟簡短。從1kg香青蘭植物中分離得到0.71g的刺槐素,轉移率較高。
本發明獲得的刺槐素純度高,達到99%。
附圖說明
下面將結合附圖和具體實施方式對本發明的上述和其他目的、特征和優點作出進一步詳細的說明。
圖1為本發明提供方法的工藝流程圖;
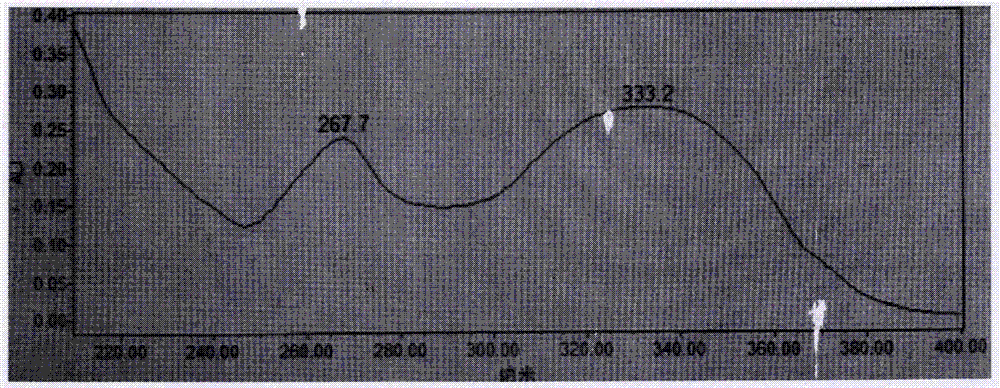
圖2為根據本發明一個實施例所制備的刺槐素的紫外光譜(UV)圖;
圖3為根據本發明一個實施例所制備的刺槐素的紅外光譜(IR)圖;
圖4為根據本發明一個實施例所制備的刺槐素的電噴霧質譜(ESI-MS)圖;
圖5為根據本發明一個實施例所制備的刺槐素的核磁共振氫譜(1H-NMR)圖;
圖6為根據本發明一個實施例所制備的刺槐素的核磁共振碳譜(13C-NMR)圖。
具體實施方式
本發明提供的一種刺槐素的制備方法,包括獲得香青蘭的提取物,以及對提取物進一步純化、層析、結晶等步驟。下面結合附圖,詳述本發明所提供的方法。
如圖1所示,首先,將1份重量的香青蘭干燥的地上部分粉碎,用15~25重量倍數的40~95vol%的醇溶液提取,例如可以選用乙醇或甲醇或丙醇或它們的任意混合物滲漉提取1~2次,每次提取6~24h。然后,將提取液濃縮,例如可以采用減壓濃縮的方法,濃縮后提取液呈膏狀。之后再將膏狀物干燥,例如可以采用真空干燥的方法,將膏狀物制成浸膏粉。
將浸膏粉用醇溶液純化,得到溶液。純化用的醇溶液包括,但不限于甲醇、乙醇、丙醇,以及它們的任意組合。優選乙醇,純化的次數一般為1~2次。
將純化后的溶液置于60℃水浴鍋中拌入聚酰胺粉,不斷攪拌直至溶劑揮干,聚酰胺拌入量為浸膏量的1/5。
將拌好樣品,進行聚酰胺柱層析。層析的步驟可以為:加適量水于拌樣聚酰胺中,使其呈混懸狀上樣。上柱完畢后,按梯度比例通入流動相進行洗脫。流動相采用復合洗脫劑,該復合洗脫劑由A組分和B組分構成,復合洗脫劑中A組分為乙醇且B組分為蒸餾水時,A組分與B組分的體積比為0∶100、2∶3、1∶1、3∶2、3∶1、11∶1。例如流動相可以采用乙醇-水或乙酸乙酯-乙醇,優選乙醇-水。此時刺槐素的組分主要在流動相乙醇-蒸餾水體積比為3∶2、3∶1時被洗脫。最后,合并相同組分洗脫液并濃縮,得到得到浸膏粉。
進一步地,為了提高刺槐素的收率,還可利用醇溶液,特別是95vol%的乙醇-水溶液,再對上述浸膏粉進行重結晶,即得到淡黃色粉末狀結晶,為刺槐素成品,其純度可達98%以上。經UV、IR、MS、1H-NMR、13C-NMR對所得組分的結構進行鑒定,可知其為刺槐素。
圖2、圖3為根據本發明的制備方法制備的刺槐素的UV圖譜和IR圖譜。
圖4為根據本發明的制備方法制備的刺槐素的ESI-MS圖譜,根據刺槐素的ESI-MS的分析可知,ESI-MS特征峰在m/z 285.20[M+](正離子),m/z 250.2[M+2OH-],m/z 302.4[M++CH3],m/z330.4[M++2OCH3-]。再結合圖5、圖6所示的根據本發明的制備方法制備的刺槐素的NMR圖譜分析,可確定其分子量為284.2,分子式為C16H12O5,其中,1H-NMR(400MHz,DMSO)圖譜中表現出典型的黃酮苷類化合物的氫譜特征,通過耦合常數及化學位移可確定每個氫的歸屬,δ:12.92(1H,S,5-OH),8.04(2H,d,J=8.8Hz,H-2′,6′),7.11(2H,d,J=8.8Hz,H-3′,5′),6.87(1H,d,H-3),6.50(1H,d,J=1.7Hz,H-8),6.20(1H,d,J=1.7Hz,H-6),3.86(3H,s,OCH3);13C-NMR(DMSO,400MHz)圖譜中呈現出16個碳信號,為1個CH3,8個CH,7個季碳,δc表明分子中含一個羰基(δ181.78),δ:163.29(C-2),103.54(C-3),181.78(C-4),164.39(C-7),162.32(C-4′),161.46(C-9),128.34(C-2′,6′),122.84(C-1′),114.60(C-3′,5′),103.71(C-10),98.95(C-8),94.07(C-6),55.57(OCH3)。因此,可確定所得化合物為刺槐素。
下面通過具體實施例對本發明作進一步詳述,以下實施例只是描述性的,不是限定性的,不能以此限定本發明的保護范圍。
實施例1:從香青蘭中提取分離得到刺槐素的方法,按以下步驟進行:
a、將香青蘭干燥的地上部分粉碎,用10倍量的40%乙醇浸泡24h;
b、采用滲漉法,香青蘭藥材3.6kg與75%乙醇比例1∶40,流速6mL/min,收集乙醇滲漉液;
c、滲漉液用孔徑0.5mm的網孔過濾2次,濾液合并;
d、用減壓蒸餾裝置,在壓力:0.08Mpa,溫度:30~60℃下,蒸餾除去乙醇得1.2g/mL的稠浸膏;水浴干燥后得浸膏粉500g;
e、將100g浸膏粉采用70%乙醇溶解后干法拌樣,上100~200目聚酰胺進行柱層析,采用40~95%乙醇梯度洗脫,得到洗脫液共90000mL,并收集60~75%乙醇梯度的洗脫液38000mL,每個梯度洗脫液為4倍量柱體積;洗脫液用減壓蒸餾裝置,在壓力:0.1Mpa,溫度:40℃下蒸干得浸膏粉10.0g;
f、為了進一步純化所得浸膏粉,濾去雜質,采用100倍量的95%乙醇對所述浸膏粉進行重結晶,所得制備液用減壓蒸餾裝置,在壓力:0.08Mpa,溫度:40℃下蒸干得淡黃色結晶0.51g。
一、結構鑒定:取所得樣品分別對UV、IR、MS、NMR進行歸峰解譜,確定實施例1所提純的樣品為刺槐素。
二、純度鑒定:
熔點(mp)測定:263℃~265℃。
薄層色譜檢測:采用兩種展開體系進行檢測,薄層板用聚酰胺薄膜。展開劑A為乙醇-甲酸水(體積比為1∶1),RfA為0.33;展開劑B為乙酸乙酯-95%乙醇(體積比為6∶4),RfB為0.61,紫外365nm下見黃色斑點,無雜質點。
HPLC檢測:用HPLC外標法測定含量,選定260nm和330nm為檢測波長。選用乙腈-0.1%磷酸水溶液(80∶20,V/V)為流動相,以刺槐素為對照品。精密稱取實施例1制備的淡黃色粉末2.50mg于100mL容量瓶,用甲醇溶解并定容至刻度線。經Waters分析型高效液相色譜檢測,外標法定量,其純度為99.12%。
得率計算:
刺槐素濃度為24.78μg·mL-1,香青蘭中刺槐素的量為2.88g/kg。
得率=(24.78μg·mL-1×100mL)/(2.50mg×3600g/2.56g×2.88g/kg)×100%=24.47%
經檢測,實施例1提取得到的刺槐素的純度為99.16%,得率為24.47%。
實施例2:從香青蘭中提取分離得到刺槐素的方法,按以下步驟進行:
a、將香青蘭干燥的地上部分粉碎,用10倍量的75%乙醇浸泡18h;
b、采用滲漉法,香青蘭藥材3.0kg與75%乙醇比例1∶35,流速6mL/min,收集乙醇滲漉液;
c、滲漉液用孔徑0.1mm的網孔過濾1次,濾液合并;
d、用減壓蒸餾裝置,在壓力:0.09Mpa,溫度:60℃下,蒸餾除去乙醇得1.0g/mL的稠浸膏;水浴干燥后得浸膏粉410g;
e、將100g浸膏粉采用70%乙醇溶解后干法拌樣,上100~200目聚酰胺進行柱層析,采用40~75%乙醇梯度洗脫得到洗脫液共75000mL,并收集60~75%乙醇梯度的洗脫液35000mL,每個梯度洗脫液為4倍量柱體積;洗脫液用減壓蒸餾裝置,在壓力:0.1Mpa,溫度:50℃下蒸干得浸膏粉9.50g;
f、為了進一步純化所得浸膏粉,濾去雜質,采用100倍量的95%乙醇對所述浸膏粉進行重結晶,所得制備液用減壓蒸餾裝置,在壓力:0.10Mpa,溫度:40℃下蒸干得淡黃色結晶0.49g。
一、結構鑒定:取所得樣品分別對UV、IR、MS、NMR進行歸峰解譜,確定實施例2所提純的樣品為刺槐素。
二、純度鑒定:
熔點(mp)測定:263℃~265℃。
薄層色譜檢測:采用兩種展開體系進行檢測,薄層板用聚酰胺薄膜。展開劑A為乙醇-甲酸水(體積比為1∶1),RfA為0.34;展開劑B為乙酸乙酯-95%乙醇(體積比為6∶4),RfB為0.61,紫外365nm下見黃色斑點,無雜質點。
HPLC檢測:用HPLC外標法測定含量,選定260nm和330nm為檢測波長。選用乙腈-0.1%磷酸水溶液(80∶20,V/V)為流動相,以刺槐素為對照品。精密稱取實施例2制備的淡黃色粉末2.50mg于100mL容量瓶,用甲醇溶解并定容至刻度線。經Waters分析型高效液相色譜檢測,外標法定量,其純度為99.36%,得率為22.85%。
實施例3:從香青蘭中提取分離得到刺槐素的方法,按以下步驟進行:
a、將香青蘭干燥的地上部分粉碎,用10倍量的40%乙醇浸泡18h;
b、采用滲漉法,香青蘭藥材4.0kg與70%乙醇比例1∶35,流速6mL/min,收集乙醇滲漉液;
c、滲漉液用孔徑0.1mm的網孔過濾2次,濾液合并;
d、用減壓蒸餾裝置,在壓力:0.1Mpa,溫度:60℃下,蒸餾除去乙醇得1.2g/mL的稠浸膏;水浴干燥后得浸膏粉550g;
e、將100g浸膏粉采用70%乙醇溶解后干法拌樣,上100~200目聚酰胺進行柱層析,采用30~75%乙醇梯度洗脫得到洗脫液共95000mL,并收集60~75%乙醇梯度的洗脫液40000mL,每個梯度洗脫液為4.5倍量柱體積;洗脫液用減壓蒸餾裝置,在壓力:0.1Mpa,溫度:50℃下蒸干得浸膏粉10.3g;
f、為了進一步純化所得浸膏粉,濾去雜質,采用100倍量的95%乙醇對所述浸膏粉進行重結晶,所得制備液用減壓蒸餾裝置,在壓力:0.10Mpa,溫度:60℃下蒸干得淡黃色結晶0.52g。
一、結構鑒定:取所得樣品分別對UV、IR、MS、NMR進行歸峰解譜,確定實施例3所提純的樣品為刺槐素。
二、純度鑒定:
熔點(mp)測定:263℃~265℃。
薄層色譜檢測:采用兩種展開體系進行檢測,薄層板用聚酰胺薄膜。展開劑A為乙醇-甲酸水(體積比為1∶1),RfA為0.34;展開劑B為乙酸乙酯-95%乙醇(體積比為6∶4),RfB為0.61,紫外365nm下見黃色斑點,無雜質點。
HPLC檢測:用HPLC外標法測定含量,選定260nm和330nm為檢測波長。選用乙腈-0.1%磷酸水溶液(80∶20,V/V)為流動相,以刺槐素為對照品。精密稱取實施例2制備的淡黃色粉末2.50mg于100mL容量瓶,用甲醇溶解并定容至刻度線。經Waters分析型高效液相色譜檢測,外標法定量,其純度為97.89%,得率為24.35%。
- 還沒有人留言評論。精彩留言會獲得點贊!