甲酰胺化合物及其鹽形式和用途的制作方法
在先申請的交叉參考本發明要求享有2007年9月12日提交的美國臨時申請第60/971,654號;2007年8月2日提交的美國臨時申請第60/953,610號;2007年8月2日提交的美國臨時申請第60/953,613號;以及2007年8月2日提交的美國臨時申請第60/953,614號的權益,通過引用將上述每篇文獻全文并入本說明書。
技術領域:
本發明涉及(2S,3R)-N-(2-((3-吡啶基)甲基)-1-氮雜雙環[2.2.2]辛-3-基)苯并呋喃-2-甲酰胺,其新的鹽形式,其制備方法,新的中間體,以及治療廣泛多種病況和障礙(包括與中樞神經系統和自主神經系統功能障礙有關的那些)的方法。
背景技術:
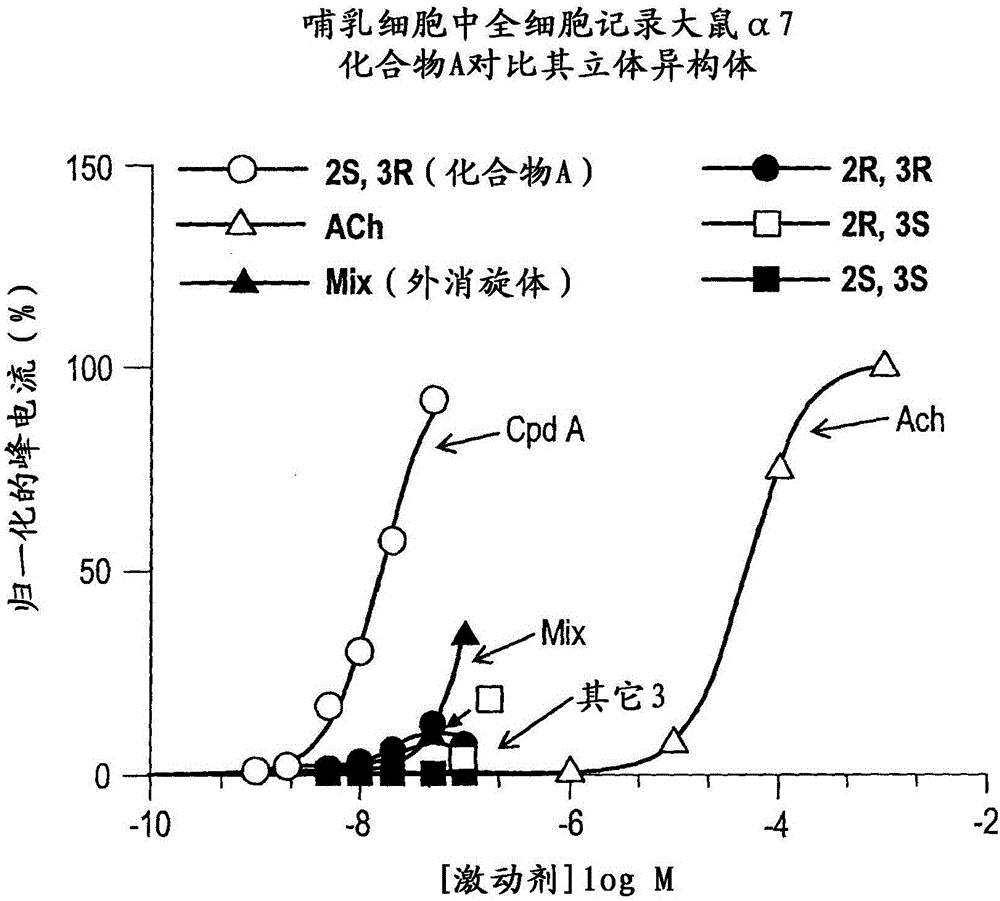
:已表明,中樞神經系統(CNS)特有的神經元煙堿樣受體(NNR)以幾種亞型存在,其中最常見的亞型是α4β2和α7亞型。例如,參見Schmitt,CurrentMed.Chem.7:749(2000),在此通過引用將其并入本說明書。與α7NNR亞型相互作用的配體已被提議可用于治療多種病況和障礙。參見Mazurov等人,Curr.Med.Chem.13:1567-1584(2006)和該文獻中引用的參考文獻,關于α7神經元煙堿樣受體亞型的背景理解,通過引用將其并入本說明書。這些病況和障礙中最為突出的有認知損傷、精神分裂癥、炎癥,血管發生,神經性疼痛和纖維肌痛。在精神分裂癥患者的尸體剖檢腦組織中,海馬NNR數目減少。而且,相對于不吸煙的精神分裂癥患者,在吸煙的精神分裂癥患者中具有改善的心理效應。煙堿改善動物和精神分裂癥患者中的感覺門控缺陷。α7NNR亞型的阻斷誘導與精神分裂癥中所見的類似的門控缺陷。例如,參見Leonard等人,SchizophreniaBulletin22(3):431(1996),在此通過引用將其并入本說明書。對在具有P50聽覺誘發電位門控缺陷的患者中的感覺加工進行的生化、分子和基因研究暗示了α7NNR亞型可能在抑制性神經元途徑中起作用。例如,參見Freedman等人,BiologicalPsychiatry38(1):22(1995),通過引用將其并入本說明書。最近,根據Heeschen等人,J.Clin.Invest.100:527(2002)所述,α7NNR被提議是血管發生的介體,通過引用將其并入本說明書。在這些研究中表明,α7亞型的抑制減少了炎性血管發生。而且,α7NNR已被提議作為控制神經發生和瘤生長的靶標(Utsugisawa等人,MolecularBrainResearch106(1-2):88(2002)和美國專利申請2002/0016371,通過引用將它們各自并入本說明書)。最后,最近認識到α7亞型在認知(Levin和Rezvani,CurrentDrugTargets:CNSandNeurologicalDisorders1(4):423(2002)),神經保護(O'Neill等人,CurrentDrugTargets:CNSandNeurologicalDisorders1(4):399(2002)以及Jeyarasasingam等人,Neuroscience109(2):275(2002)),以及神經性疼痛(Xiao等人,Proc.Nat.Acad.Sci(US)99(12):8360(2002))中的作用,通過引用將上述每篇參考文獻并入本說明書。據報道,多種化合物與α7NNR相互作用并已根據這一基礎被提議作為治療劑。例如,參見,PCTWO99/62505,PCTWO99/03859,PCTWO97/30998,PCTWO01/36417,PCTWO02/15662,PCTWO02/16355,PCTWO02/16356,PCTWO02/16357,PCTWO02/16358,PCTWO02/17358,Stevens等人,Psychopharm.136:320(1998),Dolle等人,J.LabelledComp.Radiopharm.44:785(2001)和Macor等人,Bioorg.Med.Chem.Lett.11:319(2001)和其中所引用的參考文獻,關于α7NNR和被提議的治療劑的背景教導,通過引用將上述參考文獻并入本說明書。在這些化合物中,共同的結構主題是被取代的叔雙環胺的結構(例如奎寧環)。也報道了類似的被取代的奎寧環化合物與毒蕈堿性受體結合。例如,參見Sabb的美國專利5,712,270以及PCTWO02/00652和PCTWO02/051841,關于這些化合物,通過引用將每篇參考文獻并入本說明書。一些煙堿樣化合物的限制是它們伴有各種不希望的副作用,例如通過刺激肌肉和神經節受體而引起的那些副作用。繼續需要用于預防或治療各種病況或障礙諸如CNS障礙,包括緩解這些障礙的癥狀的化合物、組合物和方法,其中所述化合物表現出具有有益效果的煙堿樣藥理學,即,影響CNS發揮作用,但無顯著的有關副作用。仍然需要影響CNS功能而不顯著影響那些可能誘導不希望的副作用諸如在心血管和骨骼肌部位處的明顯活性的煙堿樣受體亞型的化合物、組合物和方法。本發明提供了這類化合物、組合物和方法。發明概述本發明的一個方面是(2S,3R)N-(2-((3-吡啶基)甲基-1-氮雜雙環[2.2.2]辛-3-基)苯并呋喃-2-甲酰胺或其藥學上可接受的鹽。本發明的另一個方面是實質上純形式的(2S,3R)N-(2-((3-吡啶基)甲基-1-氮雜雙環[2.2.2]辛-3-基)苯并呋喃-2-甲酰胺或其藥學上可接受的鹽。本發明的另一個方面是實質上不含(2S,3S)、(2R,3S)或(2R,3R)異構體的(2S,3R)N-(2-((3-吡啶基)甲基-1-氮雜雙環[2.2.2]辛-3-基)苯并呋喃-2-甲酰胺或其藥學上可接受的鹽。另外,本發明的另一個方面是立體異構體富集的(2S,3R)-N-(2-((3-吡啶基)甲基-1-氮雜雙環[2.2.2]辛-3-基)苯并呋喃-2-甲酰胺或其藥學上可接受的鹽。在一個實施方案中,對映體和/或非對映體過量是90%或更大。在一個實施方案中,對映體和/或非對映體過量是95%或更大。在一個實施方案中,對映體和/或非對映體過量是98%或更大。在一個實施方案中,對映體和/或非對映體過量是99%或更大。在一個實施方案中,對映體和/或非對映體過量是99.5%或更大。本發明的另一個方面是(2S,3R)N-(2-((3-吡啶基)甲基-1-氮雜雙環[2.2.2]辛-3-基)苯并呋喃-2-甲酰胺的酸鹽,其中所述酸選自:鹽酸,硫酸,磷酸,馬來酸,對甲苯磺酸,半乳糖二酸(粘酸),D-扁桃酸,D-酒石酸,甲磺酸,R-和S-10-樟腦磺酸,酮戊二酸或馬尿酸。在一個實施方案中,(2S,3R)-N-(2-((3-吡啶基)甲基-1-氮雜雙環[2.2.2]辛-3-基)苯并呋喃-2-甲酰胺與酸的化學計量是2:1、1:1或1:2。在一個實施方案中,所述化學計量是1:1。本發明的一個實施方案是(2S,3R)-N-(2-((3-吡啶基)甲基)-1-氮雜雙環[2.2.2]辛-3-基)苯并呋喃-2-甲酰胺鹽酸鹽或其水合物或溶劑合物,包括部分水合物或部分溶劑合物。另一個實施方案是(2S,3R)-N-(2-((3-吡啶基)甲基)-1-氮雜雙環[2.2.2]辛-3-基)苯并呋喃-2-甲酰胺一鹽酸鹽或其水合物或溶劑合物,包括部分水合物或部分溶劑合物。本發明還提供了(2S,3R)-N-(2-((3-吡啶基)甲基)-1-氮雜雙環[2.2.2]辛-3-基)苯并呋喃-2-甲酰胺和新的中間體的可放大規模的合成。本發明的范圍包括本說明書所述的各方面、實施方案和優先選擇的所有組合。附圖說明圖1A1-1A4圖示了在哺乳動物GH4C1細胞中表達的大鼠α7受體對(2S,3R)-N-(2-((3-吡啶基)甲基)-1-氮雜雙環[2.2.2]辛-3-基)苯并呋喃-2-甲酰胺;外消旋物,即(2S,3R)、(2R,3S)、(2R,3R)和(2S,3S)的混合物;單獨的立體異構體;以及乙酰膽堿(ACh)的應答。圖1B圖示了在哺乳動物GH4C1細胞中表達的大鼠α7受體對處于有效血漿濃度范圍內的(2S,3R)-N-(2-((3-吡啶基)甲基)-1-氮雜雙環[2.2.2]辛-3-基)苯并呋喃-2-甲酰胺;外消旋物,即(2S,3R)、(2R,3S)、(2R,3R)和(2S,3S)的混合物;以及單獨的立體異構體的功能應答的比較。圖2A圖示了在非洲蟾蜍卵母細胞中表達的人α7受體對(2S,3R)-N-(2-((3-吡啶基)甲基)-1-氮雜雙環[2.2.2]辛-3-基)苯并呋喃-2-甲酰胺的應答。圖2B圖示了在施用所示濃度的化合物后人α7受體的對照應答。將數據歸一化為在實驗激動劑誘發的應答前5分鐘所獲得的對照300μMACh應答的凈電荷。每個點代表至少4個卵母細胞的歸一化應答的平均數±SEM。圖3圖示了在物體識別(OR)模型中的認知作用的評價,證明了(2S,3R)-N-(2-((3-吡啶基)甲基)-1-氮雜雙環[2.2.2]辛-3-基)苯并呋喃-2-甲酰胺以0.3和1mg/kg腹膜內給藥時具有積極影響,*p<0.5。圖4圖示了在OR模型中的認知作用的評價,證明了(2S,3R)-N-(2-((3-吡啶基)甲基)-1-氮雜雙環[2.2.2]辛-3-基)苯并呋喃-2-甲酰胺在寬的劑量范圍(0.3-10mg/kg)內口服給藥時具有積極影響,*p<0.5。圖5圖示了腹膜內給藥的(2S,3R)-N-(2-((3-吡啶基)甲基)-1-氮雜雙環[2.2.2]辛-3-基)苯并呋喃-2-甲酰胺在OR任務中在預防由MK-801(亦稱地佐環平,一種市售的NMDA受體的非競爭性拮抗劑)誘導的認知缺陷中的作用。圖6圖示了在OR任務中由用介質處理的組在最后的亞急性給藥(經口給藥)后30分鐘、6小時或24小時在物體A上所花費的平均時間相對于在物體B上所花費的平均時間沒有顯著差別(分別是p=0.17,p=0.35和p=0.12)。或者,在最后的亞急性給藥0.3mg/kg的(2S,3R)-N-(2-((3-吡啶基)甲基)-1-氮雜雙環[2.2.2]辛-3-基)苯并呋喃-2-甲酰胺后30分鐘、2小時、6小時和18小時,受試者探索物體B(新的物體)比探索物體A(熟悉的物體)花費顯著(P<0.05)更多的時間。另外,在用0.3mg/kg的(2S,3R)-N-(2-((3-吡啶基)甲基)-1-氮雜雙環[2.2.2]辛-3-基)苯并呋喃-2-甲酰胺處理的動物中的認知指數在2小時(75%)和6小時(71%)顯著高于在最后給藥后30分鐘時用介質處理的組的認知指數(54%)。圖7圖示了在放射狀臂形迷宮(RAM)模型中的認知作用的評價。在每日訓練期間之前30分鐘口服給藥(2S,3R)-N-(2-((3-吡啶基)甲基)-1-氮雜雙環[2.2.2]辛-3-基)苯并呋喃-2-甲酰胺(0.1、0.3和1.0mg/kg)。在給藥的第二周期間,在用0.3mg/kg的(2S,3R)-N-(2-((3-吡啶基)甲基)-1-氮雜雙環[2.2.2]辛-3-基)苯并呋喃-2-甲酰胺處理的組中,任務的行為表現的改善是明顯的。圖8圖示了作為由多巴胺過度刺激所誘導的活動過強行為被測量的抗精神病效果的研究,表明(2S,3R)-N-(2-((3-吡啶基)甲基)-1-氮雜雙環[2.2.2]辛-3-基)苯并呋喃-2-甲酰胺(0.3和1.0mg/kg;皮下給藥)在大鼠中皮下給藥后減弱由阿撲嗎啡(1.0mg/kg)所誘導的運動活性過強。圖9圖示了抗精神病評價,前脈沖抑制,顯示由阿撲嗎啡誘導的缺陷在皮下給藥后被(2S,3R)-N-(2-((3-吡啶基)甲基)-1-氮雜雙環[2.2.2]辛-3-基)苯并呋喃-2-甲酰胺的預處理所逆轉。圖10A圖示了(2S,3R)-N-(2-((3-吡啶基)甲基)-1-氮雜雙環[2.2.2]辛-3-基)苯并呋喃-2-甲酰胺一鹽酸鹽的X射線結晶學分析的結果,確認了該物質的絕對立體化學。被描繪的化合物是部分水合的鹽酸鹽,完全有序的氯離子和在不對稱單元中部分占據的水分子顯示了這一點。圖10B圖示了(2S,3R)-N-(2-((3-吡啶基)甲基)-1-氮雜雙環[2.2.2]辛-3-基)苯并呋喃-2-甲酰胺一鹽酸鹽的X射線結晶學分析的結果,確認了該物質的絕對立體化學,使用編號方案描述供參考。視圖是向下觀察晶胞的結晶學b-軸。分子之間的氫鍵用虛線表示。圖11A圖示了(2R,3R)-N-(2-((3-吡啶基)甲基)-1-氮雜雙環[2.2.2]辛-3-基)苯并呋喃-2-甲酰胺對氯苯甲酸鹽的X射線結晶學分析的結果,確認了該物質的絕對立體化學。圖11B圖示了(2R,3R)-N-(2-((3-吡啶基)甲基)-1-氮雜雙環[2.2.2]辛-3-基)苯并呋喃-2-甲酰胺對氯苯甲酸鹽的X射線結晶學分析的結果,確立了該物質的絕對立體化學,使用編號方案描述供參考。圖12圖示了表征N-(2-((3-吡啶基)甲基)-1-氮雜雙環[2.2.2]辛-3-基)苯并呋喃-2-甲酰胺的四種立體異構體的完整色譜圖,其中2S,3R在5.3分鐘保留時間處顯示出峰,2R,3S在7.3分鐘的保留時間處顯示出峰,2R,3R在8.2分鐘的保留時間處顯示出峰,以及2S,3S在12.4分鐘的保留時間處顯示出峰。如本說明書中所述,分析以提供了足夠的分辨率所需的流動相形成60:40:0.2的己烷:乙醇:二正丁胺的組合物,流速為1.0毫升/分鐘,柱溫為20℃,以及UV檢測波長是270nm。圖13是(2S,3R)-N-(2-((3-吡啶基)甲基)-1-氮雜雙環[2.2.2]辛-3-基)苯并呋喃-2-甲酰胺一鹽酸鹽的XRPD,圖示了觀察圖形(較亮)和計算圖形(較暗)。兩個圖形在2θ值方面是一致的,在強度和峰寬方面的較小差別可歸于儀器分辨率和優選取向效應。如本說明書中所述,其它較小差異可歸因于由于在室溫下被收集的觀察數據和從120K的結構計算的數據所導致的溫度變動。圖14是(2S,3R)-N-(2-((3-吡啶基)甲基)-1-氮雜雙環[2.2.2]辛-3-基)苯并呋喃-2-甲酰胺一甲苯磺酸鹽的XRPD。發明的詳細說明對α7NNR亞型具有親合力(≤1nMKi值)和選擇性的具體化合物(2S,3R)-N-(2-((3-吡啶基)甲基)-1-氮雜雙環[2.2.2]辛-3-基)苯并呋喃-2-甲酰胺在認知(認知增強)和精神病(抗精神病效果)的動物模型中顯示了功效。本發明的一個方面是(2S,3R)-N-(2-((3-吡啶基)甲基-1-氮雜雙環[2.2.2]辛-3-基)苯并呋喃-2-甲酰胺或其藥學上可接受的鹽。另一個方面是實質上純形式的(2S,3R)-N-(2-((3-吡啶基)甲基-1-氮雜雙環[2.2.2]辛-3-基)苯并呋喃-2-甲酰胺或其藥學上可接受的鹽。另一個方面是實質上不含(2S,3S)、(2R,3S)或(2R,3R)異構體的(2S,3R)N-(2-((3-吡啶基)甲基-1-氮雜雙環[2.2.2]辛-3-基)苯并呋喃-2-甲酰胺或其藥學上可接受的鹽。另外,另一個方面是立體異構體富集的(2S,3R)-N-(2-((3-吡啶基)甲基-1-氮雜雙環[2.2.2]辛-3-基)苯并呋喃-2-甲酰胺或其藥學上可接受的鹽。在一個實施方案中,對映體和/或非對映體過量是90%或更大。在一個實施方案中,對映體和/或非對映體過量是95%或更大。在一個實施方案中,對映體和/或非對映體過量是98%或更大。在一個實施方案中,對映體和/或非對映體過量99%或更大。在一個實施方案中,對映體和/或非對映體過量是99.5%或更大。本發明的另一個方面是(2S,3R)N-(2-((3-吡啶基)甲基-1-氮雜雙環[2.2.2]辛-3-基)苯并呋喃-2-甲酰胺的酸鹽,其中所述酸選自:鹽酸,硫酸,磷酸,馬來酸,對甲苯磺酸,半乳糖二酸(粘酸),D-扁桃酸,D-酒石酸,甲磺酸,R-和S-10-樟腦磺酸,酮戊二酸或馬尿酸。在一個實施方案中,(2S,3R)-N-(2-((3-吡啶基)甲基-1-氮雜雙環[2.2.2]辛-3-基)苯并呋喃-2-甲酰胺與酸的化學計量是2:1、1:1或1:2。在一個實施方案中,所述化學計量是1:1。本發明的一個實施方案是(2S,3R)-N-(2-((3-吡啶基)甲基)-1-氮雜雙環[2.2.2]辛-3-基)苯并呋喃-2-甲酰胺鹽酸鹽或其水合物或溶劑合物,包括部分水合物或部分溶劑合物。另一個實施方案是(2S,3R)-N-(2-((3-吡啶基)甲基)-1-氮雜雙環[2.2.2]辛-3-基)苯并呋喃-2-甲酰胺一鹽酸鹽或其水合物或溶劑合物,包括部分水合物或部分溶劑合物。本發明的另一個方面是(2S,3R)-(2-((3-吡啶基)甲基)-3-氨基-1-氮雜雙環[2.2.2]辛烷。本發明的另一個方面是包含本發明的化合物和一種或多種藥學上可接受的載體的藥物組合物。本發明的另一個方面是治療或預防中樞神經系統障礙、炎癥、疼痛或新血管形成方法,它包括給予本發明的化合物。在一個實施方案中,所述中樞神經系統障礙以正常神經遞質釋放的改變為特征。在一個實施方案中,所述中樞神經系統障礙選自輕度認知損傷、年齡相關的記憶損傷、早老性癡呆、早發性阿爾茨海默病、老年性癡呆、阿爾茨海默病型癡呆、阿爾茨海默病、萊維體(LewyBody)癡呆、微梗塞性癡呆、AIDS相關性癡呆、HIV癡呆、多發性腦梗塞、帕金森神經功能障礙、帕金森病、皮克病、進行性核上性麻痹、亨廷頓舞蹈病、遲發性運動障礙、運動機能亢進、躁狂癥、注意缺陷障礙、注意渙散多動癥、焦慮癥、抑郁癥、誦讀困難、精神分裂癥、精神分裂癥中的認知功能障礙、抑郁癥、強迫觀念與行為障礙或圖雷特綜合征。在一個實施方案中,所述中樞神經系統障礙選自阿爾茨海默病、躁狂癥、注意缺陷障礙、注意渙散多動癥、焦慮癥、誦讀困難、精神分裂癥、精神分裂癥中的認知功能障礙、抑郁癥、強迫觀念與行為障礙或圖雷特綜合征。本發明的另一個方面包括本發明的化合物在制備用于治療或預防中樞神經系統障礙、炎癥、疼痛或新血管形成的藥物中的用途。在一個實施方案中,所述中樞神經系統障礙以正常神經遞質釋放的改變為特征。在一個實施方案中,所述中樞神經系統障礙選自輕度認知損傷、年齡相關的記憶損傷、早老性癡呆、早發性阿爾茨海默病、老年性癡呆、阿爾茨海默病型癡呆、阿爾茨海默病、萊維小體癡呆、微梗塞性癡呆、AIDS相關性癡呆、HIV癡呆、多發性腦梗塞、帕金森神經功能障礙、帕金森病、皮克病、進行性核上性麻痹、亨廷頓舞蹈病、遲發性運動障礙、運動機能亢進、躁狂癥、注意缺陷障礙、注意渙散多動癥、焦慮癥、抑郁癥、誦讀困難、精神分裂癥、精神分裂癥中的認知功能障礙、抑郁癥、強迫觀念與行為障礙或圖雷特綜合征。在一個實施方案中,所述中樞神經系統障礙選自阿爾茨海默病、躁狂癥、注意缺陷障礙、注意渙散多動癥、焦慮癥、誦讀困難、精神分裂癥、精神分裂癥中的認知功能障礙、抑郁癥、強迫觀念與行為障礙或圖雷特綜合征。本發明的另一個方面是用于治療或預防中樞神經系統障礙、炎癥、疼痛或新血管形成的本發明的化合物。在一個實施方案中,所述中樞神經系統障礙以正常神經遞質釋放的改變為特征。在一個實施方案中,所述中樞神經系統障礙選自輕度認知損傷、年齡相關的記憶損傷、早老性癡呆、早發性阿爾茨海默病、老年性癡呆、阿爾茨海默病型癡呆、阿爾茨海默病、萊維小體癡呆、微梗塞性癡呆、AIDS相關性癡呆、HIV癡呆、多發性腦梗塞、帕金森神經功能障礙、帕金森病、皮克病、進行性核上性麻痹、亨廷頓舞蹈病、遲發性運動障礙、運動機能亢進、躁狂癥、注意缺陷障礙、注意渙散多動癥、焦慮癥、抑郁癥、誦讀困難、精神分裂癥、精神分裂癥中的認知功能障礙、抑郁癥、強迫觀念與行為障礙或圖雷特綜合征。在一個實施方案中,所述中樞神經系統障礙選自阿爾茨海默病、躁狂癥、注意缺陷障礙、注意渙散多動癥、焦慮癥、誦讀困難、精神分裂癥、精神分裂癥中的認知功能障礙、抑郁癥、強迫觀念與行為障礙或圖雷特綜合征。在上述的方法和用途中,在本發明的一個實施方案中,有效劑量為每24小時期間約1毫克至10毫克。本發明的另一個方面是通過(2-((3-吡啶基)甲基-1-氮雜雙環[2.2.2]辛-3-酮的依次動力學拆分和立體選擇性還原胺化,制備(2S,3R)-N-(2-((3-吡啶基)甲基-1-氮雜雙環[2.2.2]辛-3-基)苯并呋喃-2-甲酰胺或其藥學上可接受的鹽的方法。本發明的范圍包括本說明書所述的各方面、實施方案和優先選擇的所有組合。藥物候選物的商業性開發涉及許多步驟,包括按比例放大化學合成和純化,發現最佳的鹽形式,等等。在藥物組合物的制劑中,藥物物質優選處于可方便地進行處理和加工的形式。需要考慮的事項包括商業可行性以及制造的一致性。另外,在制備藥物組合物中,重要的是在給藥至患者后,提供可靠的、可重現的和恒定的藥物血漿濃度曲線。活性成分的化學穩定性、固態穩定性和“儲存期限”也是非常重要的因素。這些藥物物質和包含這些藥物物質的組合物應優選能夠在可估計的時段內被有效保存,在活性組分的物化特征方面(例如,其化學組成,密度、吸濕性和溶解性)不表現出重大變化。另外,同樣重要的是能夠提供盡可能化學純的形式的藥物。在下文中更詳細地討論本發明的這些特征。I.化合物本發明的化合物是由以下化合物A表示的(2S,3R)-N(2-((3-吡啶基)甲基)-1-氮雜雙環[2.2.2]辛-3-基)苯并呋喃-2-甲酰胺或化合物A的藥學上可接受的鹽的形式。在Mazurov等人的美國專利No.6,953,855中描述了外消旋化合物N-(2-((3-吡啶基)甲基-1-氮雜雙環[2.2.2]辛-3-基)苯并呋喃-2-甲酰胺、合成和醫學治療的效用,在此通過引用將其并入本說明書。外消旋的N-(2-((3-吡啶基)甲基-1-氮雜雙環[2.2.2]辛-3-基)苯并呋喃-2-甲酰胺是神經元煙堿樣受體(NNR)的α7亞型的高親和力配體。外消旋的N-(2-((3-吡啶基)甲基-1-氮雜雙環[2.2.2]辛-3-基)苯并呋喃-2-甲酰胺含有兩個不對稱取代的碳原子。因此,外消旋的N-(2-((3-吡啶基)甲基-1-氮雜雙環[2.2.2]辛-3-基)苯并呋喃-2-甲酰胺以四種立體異構體形式即(S,S)、(S,R)、(R,R)和(R,S)存在。(S,R),即(2S,3R),是化合物A。先前認為,在報道的合成(包括美國專利6,953,855)中產生的主要的立體異構形式以在1-氮雜雙環[2.2.2]辛烷(奎寧環)環的2和3位處的順式相對構型為特征。換句話說,認為順式非對映體(對映體的(2R,3R)和(2S,3S)對)是根據報道的方法制備時得到的主要形式。這一主要順式合成的確定基于:(i)奎寧環以及分離的非對映(順式和反式)中間體的2位和3位氫核的1H偶合常數與文獻報道的偶合常數的比較;以及(ii)通過類似于文獻所述,所述文獻如Warawa等人,J.Med.Chem.18(6):587-593(1975)和Viti等人,LetrahedronLett.35(32):5939-5942(1994),兩篇文獻都通過引用將其并入本說明書,用于產生化合物混合物的合成化學的預期立體化學結果。因此,預料到將形成順式構型。就這點而論,使用外消旋物進行生物試驗所產生的結果被假定為歸因于主要的順式構型。目前借助于結晶鹽形式和類似物的X射線衍射分析已經發現,在最初合成中產生的主要的非對映體實際上是反式非對映體。另外,已經發現具有反式相對立體化學的兩種對映體即(2S,3R)和(2R,3S),在它們與α7NNR亞型相互作用的能力方面彼此實質上不同。(2S,3R)構型,即化合物A,具有更大的活性。通過進一步的分析,已經發現化合物A具有使其與:i)單獨獲得的其它三種立體異構體中的每一種,ii)所有四種立體異構體的混合物,即外消旋物;以及iii)在文獻中報道的其它α7NNR配體相區別的藥理學性質。(2S,3R)-N(2-((3-吡啶基)甲基)-1-氮雜雙環[2.2.2]辛-3-基)苯并呋喃-2-甲酰胺(化合物A)是在α7NNR受體處的高度選擇性的完全激動劑,其具有顯著低的EC50(用于激活)值并且在EC50和IC50(用于殘余抑制)之間具有良好的距離差,在寬的治療可用濃度范圍內提供了功能激動。II.(2S,3R)-N-(2-((3-吡啶基)甲基)-1-氮雜雙環[2.2.2]辛-3-基)苯并呋喃-2-甲酰胺的可放大規模合成具體的合成步驟在它們按比例放大的可行性方面有差異。發現由于多種原因而使得反應缺乏按比例放大的能力,所述原因包括安全考慮、試劑費用、困難的后處理或純化、反應能學(熱力學或動力學)和反應收率。本說明書所述的(2S,3R)-N-(2-((3-吡啶基)甲基-1-氮雜雙環[2.2.2]辛-3-基)苯并呋喃-2-甲酰胺的合成已用于生成千克量級的材料,并且已經以高收率在數千克規模上進行了組分反應。可按比例放大的合成采用了可外消旋的酮(2-((3-吡啶基)甲基)-1-氮雜雙環[2.2.2]辛烷-3-酮)的動態拆分和經過拆分的酮的(R)-α-甲基芐基胺亞胺衍生物的立體選擇性還原(還原胺化)。本說明書所報道的合成順序可容易地進行規模放大并免除色譜純化。III.(2S,3R)-N-(2-((3-吡啶基)甲基-1-氮雜雙環[2.2.2]辛-3-基)苯并呋喃-2-甲酰胺的新的鹽形式的制備(2S,3R)-N-(2-((3-吡啶基)甲基-1-氮雜雙環[2.2.2]辛-3-基)苯并呋喃-2-甲酰胺作為游離堿是具有非常有限的水溶性的無定形粉末。該游離堿與無機酸和有機酸反應以生成某些酸加成鹽,這些酸加成鹽具有對于藥物組合物的制備而言是有利的物理和化學性質,包括但不限于結晶度、水溶性和穩定性。本發明的鹽的化學計量可以改變。根據形成本說明書所述鹽的方式,鹽可具有吸留了在成鹽期間所存在的溶劑的晶體結構。因此,鹽可以作為相對于(2S,3R)-N-(2-((3-吡啶基)甲基-1-氮雜雙環[2.2.2]辛-3-基)苯并呋喃-2-甲酰胺具有不同化學計量的溶劑的水合物和其它溶劑合物存在。制備鹽形式的方法可以改變。(2S,3R)-N-(2-((3-吡啶基)甲基-1-氮雜雙環[2.2.2]辛-3-基)苯并呋喃-2-甲酰胺鹽形式的制備一般包括:(i)將游離堿或游離堿的溶液即(2S,3R)-N-(2-((3-吡啶基)甲基-1-氮雜雙環[2.2.2]辛-3-基)苯并呋喃-2-甲酰胺在合適的溶劑中的溶液與純酸混合,或者與該酸在合適的溶劑中的溶液混合;(iia)如有必要,將得到的鹽溶液冷卻,以引起沉淀;或(iib)加入合適的抗溶劑以引起沉淀;或(iic)蒸發第一溶劑并加入新的溶劑并重復步驟(iia)或步驟(iib);以及(iii)過濾和收集得到的鹽。在一個實施方案中,(2S,3R)-N-(2-((3-吡啶基)甲基-1-氮雜雙環[2.2.2]辛-3-基)苯并呋喃-2-甲酰胺是立體異構體富集的。在一個實施方案中,對映體和/或非對映體過量是90%或更大。在一個實施方案中,對映體和/或非對映體過量是95%或更大。在一個實施方案中,對映體和/或非對映體過量是98%或更大。在一個實施方案中,對映體和/或非對映體過量是99%或更大。在一個實施方案中,對映體和/或非對映體過量是99.5%或更大。使用的化學計量、溶劑配合比、溶質濃度和溫度可以改變。可用于鹽形式的制備或重結晶的代表性溶劑包括但不限于乙醇、甲醇、異丙醇、丙酮、乙酸乙酯和乙腈。合適的藥學上可接受的鹽的實例包括無機酸加成鹽諸如氯化物,溴化物,硫酸鹽,磷酸鹽和硝酸鹽;有機酸加成鹽諸如乙酸鹽,半乳糖二酸鹽,丙酸鹽,琥珀酸鹽,乳酸鹽,羥乙酸鹽,蘋果酸鹽,酒石酸鹽,檸檬酸鹽,馬來酸鹽,富馬酸鹽,甲磺酸鹽,對甲苯磺酸鹽和抗壞血酸鹽;以及與氨基酸形成的鹽諸如天冬氨酸鹽和谷氨酸鹽。鹽有時候可為水合物或乙醇溶劑合物。代表性的鹽根據Dull等人的美國專利第5,597,919號,Dull等人的美國專利第5,616,716號和Ruecroft等人的美國專利第5,663,356號所述被提供,通過引用將它們各自并入本說明書。對于游離堿(2S,3R)-N-(2-((3-吡啶基)甲基-1-氮雜雙環[2.2.2]辛-3-基)苯并呋喃-2-甲酰胺進行的鹽篩選顯示,盡管可形成許多的藥學上可接受的酸的鹽,但是這些鹽中只有少數具有對于商業制造而言是可接受的性質。因此,不存在預測由商業可行的鹽所體現的特征的能力。提供作為結晶的鹽(即顯示一定的結晶度的鹽)的酸,根據制備鹽方法,包括鹽酸,硫酸,磷酸,對甲苯磺酸,半乳糖二酸(粘酸),D-扁桃酸,D-酒石酸,甲磺酸,R-和S-10-樟腦磺酸,馬來酸,酮戊二酸和馬尿酸。在這些鹽中,鹽酸、磷酸、馬來酸和對甲苯磺酸的鹽各自表現出另外的合乎需要的性質,包括高熔點、良好的水溶性和低吸濕性。這些鹽中的這些特征是意想不到的。IV.藥物組合物本發明的藥物組合物包括本說明書所述的鹽,所述鹽為純態或為其中所述化合物與任何其它藥學相容的產品(其可為惰性的或具生理學活性的)相組合的組合物形式。得到的藥物組合物可用于預防易患病況或障礙的受試者中的所述病況或障礙,以及/或治療罹患所述病況或障礙的受試者。本說明書的藥物組合物包括本發明的化合物和/或其藥學上可接受的鹽。化合物的給藥方式可以改變。所述組合物優選口服給藥(例如,在溶劑諸如水性或非水性液體中的液體形式中,或在固體載體中)。用于口服給藥的優選組合物包括丸劑,片劑,膠囊,膠囊性片劑,糖漿劑和溶液,包括硬明膠膠囊和時間釋放膠囊。標準的賦形劑包括粘合劑、填充劑、著色劑、增溶劑等等。組合物可被配制成單位劑量形式,或為多劑量或亞單位劑量。優選的組合物為液體或半固體形式。可以使用包括液體藥學惰性載體諸如水或其它藥學相容的液體或半固體的組合物。這些液體和半固體的使用是本領域技術人員公知的。組合物還可通過注射即靜脈內、肌內、皮下、腹膜內、動脈內、鞘內;以及腦室內途徑給藥。靜脈內給藥是優選的注射方法。合適的注射用載體是本領域技術人員公知的并且包括5%的葡萄糖溶液、鹽水和磷酸鹽緩沖鹽水。藥物產品還可作為輸注劑或注射劑給予(例如作為在藥學上可接受的液體或液體混合物中的懸浮液或乳液)。制劑還可使用其它手段例如直腸給藥進行給藥。可用于直腸給藥的制劑諸如栓劑是本領域技術人員公知的。藥物產品還可通過吸入(例如,以氣霧劑形式經鼻或使用Brooks等人的美國專利第4,922,901號中所述類型的遞送制品進行,將該文獻全文并入本說明書);局部(例如,以洗劑形式);透皮(例如,使用透皮貼劑)或離子導入方式給藥;或通過舌下或經頰給藥。盡管有可能給予呈本體活性化學品形式的化合物,但是優選提供用于有效率的和有功效的給藥的藥物組合物或藥物制劑形式的藥物產品。化合物給藥的示例性方法是本領域技術人員公知的。這些制劑的有用性可根據所用的具體組合物和接受治療的具體受試者的不同而異。這些制劑可含有液體載體,該液體載體可以是油性的、水性的、被乳化的或含有某些適合于給藥方式的溶劑。組合物可以周期性地或以漸變的、連續的、恒定的或受控的速率被給予至溫血動物(例如哺乳動物,諸如小鼠,大鼠,貓,兔,狗,豬,牛或猴),但是有利地被給予至人類。另外,藥物制劑的給藥天數和每天給藥次數可以改變。用于本發明化合物給藥的其它合適的方法描述于Smith等人美國專利第5,604,231號中,通過引用將該文獻的內容并入本說明書。在本發明的實施方案中并且可由本領域技術人員所理解的,本發明的化合物可與其它的治療化合物組合給藥。例如,本發明的化合物可與諸如以下的物質組合使用:其他的NNR配體(諸如伐尼克蘭),抗氧化劑(諸如自由基清除劑),抗細菌藥(諸如青霉素類抗生素),抗病毒藥(諸如核苷類似物,如齊多夫定和阿昔洛韋),抗凝血藥(諸如華法林),抗炎藥(諸如NSAID),解熱藥,鎮痛藥,麻醉藥(諸如手術中使用的麻醉藥),乙酰膽堿酯酶抑制劑(諸如多奈哌齊和加蘭他敏),抗精神病藥(諸如氟哌啶醇,氯氮平,奧氮平和喹硫平),免疫抑制劑(諸如環孢菌素和甲氨蝶呤),神經保護藥,甾族化合物(諸如甾體激素),皮質類固醇(諸如地塞米松,強的松和氫化可的松),維生素,礦物質,營養制品,抗抑郁藥(諸如丙米嗪,氟西汀,帕羅西汀,依他普侖,舍曲林,文拉法辛和度洛西汀),抗焦慮藥(諸如阿普唑侖和丁螺環酮),抗驚厥藥(諸如苯妥英和加巴噴丁),血管擴張藥(諸如哌唑嗪和西地那非),情緒穩定藥(諸如丙戊酸鹽和阿立哌唑),抗癌藥(諸如抗增殖藥),抗高血壓藥(諸如阿替洛爾,可樂定,氨氯地平,維拉帕米和奧美沙坦),輕瀉藥,糞便軟化藥,利尿藥(諸如呋塞米),解痙藥(諸如雙環維林),抗運動障礙藥和抗潰瘍藥(諸如艾美拉唑)。本發明的化合物可單獨使用或與其它的治療劑組合使用。藥學活性劑的這種組合可以一起或分開進行給藥,并且當分開給藥時,給藥可以同時進行或以任何順序依次進行。化合物或藥劑的量以及給藥的相對時機將經過選擇,以便獲得所需的治療效果。組合給藥可以是以下形式的同時給藥:(1)包括多種化合物的整體式藥物組合物;或(2)單獨的藥物組合物,其各自包含所述化合物中的一種。或者,所述組合可以順序方式分開給藥,在該順序方式中,先給予一種治療劑,然后給予另一種治療劑,或者反之亦然。該順序給藥在時間上可以間隔較近或較遠。本發明的化合物可用于治療多種障礙和病況,并且因此,本發明的化合物可與多種其它合適的可用于治療或預防這些障礙或病況的治療劑組合使用。化合物的合適的劑量是有效防止罹患障礙的患者發生所述障礙的癥狀或治療所述障礙的一些癥狀的量。所提到的“有效量”、“治療量”或“有效劑量”是指足以引發所需的藥理學或治療學效果從而導致有效預防或治療障礙的量。當治療CNS障礙時,化合物的有效量是足以穿過受試者的血腦屏障以便結合至該受試者的腦中的有關受體部位并調節有關NNR亞型的活性(例如提供神經遞質分泌,從而導致有效預防或治療障礙)的量。障礙的預防的實例體現為延遲障礙的癥狀的發生。障礙的治療的實例體現為減少與障礙有關的癥狀或改善障礙癥狀的復發。優選地,有效量足以獲得所需的結果,但不足以引起明顯的副作用。有效劑量可根據諸如患者的狀況、障礙的癥狀的嚴重程度和藥物組合物的給藥方式的因素不同而異。對于人類患者,典型化合物的有效劑量一般要求給予一定量的化合物,該量足以調節有關的NNR的活性,但是該量應不足以誘導對骨骼肌和神經節的影響達到任何顯著程度。化合物的有效劑量當然因患者與患者之間的不同而異,但是通常包括從以發生CNS效果或其它所需治療效果為起點、但低于觀察到肌肉效果的量的范圍內的量。本說明書所述的化合物,當根據本說明書所述方法以有效量被使用時,可以提供一定程度的對CNS障礙或其它障礙的進展的預防、CNS障礙或其它障礙的癥狀的改善,或使CNS障礙或其它障礙的復發改善達到一定程度。這些化合物的有效量通常低于引發任何明顯的副作用(例如,與骨骼肌或神經節有關的那些副作用)所需的閾值濃度。化合物可在某些CNS障礙和其它障礙被治療并且某些副作用可被避免的治療窗內進行給藥。理想地是,本說明書所述化合物的有效劑量足以為障礙提供所希望的效果但是不足以(即,不在足夠高的水平下)提供不希望的副作用。優選地,化合物在有效治療CNS障礙或其它障礙但是低于引發某些副作用到任何顯著程度所需的量的1/5、通常低于1/10的劑量下被給藥。最優選地,有效劑量為當觀察到發生最大效果并具有最小副作用時的極低濃度。典型地,這些化合物的有效劑量一般要求以低于5mg/kg患者重量的量給予化合物。通常,本發明的化合物以低于約1mg/kg患者體重被給予,通常以低于約100μg/kg患者重量被給予,但是經常以約10μg到低于100μg/kg患者重量被給予。上述的有效劑量一般地代表作為單劑量被給予的量,或作為在24小時期間以一個或多個劑量被給予的量。對于人類患者,典型化合物的有效劑量一般要求以至少約1毫克/24小時/患者、通常至少約10毫克/24小時/患者、并且經常至少約100毫克/24小時/患者的量給予化合物。對于人類患者,典型化合物的有效劑量要求給予一般不超過約500毫克/24小時/患者、通常不超過約400毫克/24小時/患者、并且經常不超過約300毫克/24小時/患者的量的化合物。另外,組合物可有利地以有效劑量被給予,從而使得化合物在患者血漿內的濃度通常不超過50ng/mL,通常不超過30ng/mL,并且經常不超過10ng/mL。在本發明的一個實施方案中,在24小時期間的有效劑量為約1mg到10mg。IV.藥物組合物的用法本說明書使用的“激動劑”是刺激其結合配對物(通常是受體)的物質。刺激在具體試驗的背景下進行定義,或者根據文獻討論而是清楚明白的,其在本領域技術人員可理解的實質上類似的環境下進行了被接受為具體結合配對物的“激動劑”或“拮抗劑”的因子或物質的比較。刺激可根據由激動劑或部分激動劑與結合配對物的相互作用所誘導的具體效果或功能的增加來定義并且可包括變構作用。本說明書使用的“拮抗劑”是抑制其結合配對物(通常是受體)的物質。抑制在具體試驗的背景下進行定義,或者根據文獻討論而是清楚明白的,其在本領域技術人員可理解的實質上類似的環境下進行了被接受為具體結合配對物的“激動劑”或“拮抗劑”的因子或物質的比較。抑制可根據由拮抗劑與結合配對物的相互作用所誘導的具體效果或功能的降低來定義并且可包括變構作用。本說明書使用的“部分激動劑”或“部分拮抗劑”分別是為其結合配對物分別提供非全部的或不完全的激動或拮抗效果的刺激或抑制水平。可認識到,抑制以及由此的抑制被固有地定義用于要被定義為激動劑、拮抗劑或部分激動劑的任何物質或任何類別的物質。本說明書使用的“固有活性”或“功效”涉及結合配對物復合物的生物有效性的一些量度。關于受體藥理學,固有活性或功效被定義的背景應當根據結合配對物(例如受體/配體)復合物的背景和與特定的生物結果有關的活性的考慮而定。例如,在一些情況下,固有活性可根據所涉及的特定的第二信使系統的不同而異。參見Hoyer,D.和Boddeke,H.,TrendsPharmacol.Sci14(7):270-5(1993),關于這一教導,通過引用將其并入本說明書。這些前后關聯的具體評價在什么情況下相關聯以及它們在本發明的背景下如何相關聯,是本領域技術人員顯而易見的。本說明書使用的受體調節包括受體的激動,部分激動,拮抗,部分拮抗或反向激動。本說明書使用的其釋放受到本說明書所述化合物介導的神經遞質包括但不限于乙酰膽堿,多巴胺,去甲腎上腺素,5-羥色胺和谷氨酸,并且本說明書所述的化合物起到CNSNNR的α7亞型的調節劑的作用。本說明書使用的術語“預防”或“防止”包括疾病、障礙或病況進展的任何程度的減少,或疾病、障礙或病況發病的任何程度的延遲。該術語包括提供了對抗具體的疾病、障礙或病況的保護效果以及包括疾病、障礙或病況的復發的改善。因此,在另一個方面,本發明提供了治療患有NNR或nAChR介導的障礙的患者或者有發展或經歷由NNR或nAChR介導的障礙的復發危險的受試者的方法。本發明的化合物和藥物組合物可用于實現有益的治療或預防效果,例如,在罹患CNS功能障礙的受試者中。如上所述,本發明的游離堿和鹽化合物調節CNS所特有的α7NNR亞型,并且可通過調節α7NNR用于預防或治療罹患或易患多種病況或障礙的受試者中的該病況或障礙,包括CNS病況或障礙。化合物能夠選擇性結合于α7NNR并表達煙堿性藥理學,例如,如所述的那樣充當激動劑、部分激動劑、拮抗劑。例如,本發明的化合物當以有效量被給予至有需要的患者時,提供了CNS障礙進展的一定程度的預防,即,提供了保護作用,改善了CNS障礙的癥狀,或改善了CNS障礙的復發,或其組合。本發明的化合物可用于治療或預防其它類型的煙堿化合物已被提議或顯示可用作治療劑的那些類型的病況和障礙。例如,參見上文所列舉的文獻,以及Williams等人,DrugNewsPerspec.7(4):205(1994),Arneric等人,CNSDrugRev.1(1):1-26(1995),Arneric等人,Exp.Opin.Invest.Drugs5(1):79-100(1996),Bencherif等人,J.Pharmacol.Exp.Ther.279:1413(1996),Lippiello等人,J.Pharmacol.Exp.Ther.279:1422(1996),Damaj等人,J.Pharmacol.Exp.Ther.291:390(1999);Chiari等人,Anesthesiology91:1447(1999),Lavand'hommeandEisenbach,Anesthesiology91:1455(1999),Holladay等人,J.Med.Chem.40(28):4169-94(1997),Bannon等人,Science279:77(1998),PCTWO94/08992,PCTWO96/31475,PCTWO96/40682,以及Bencherif等人的美國專利第5,583,140號,Dull等人的美國專利第5,597,919號,Smith等人的美國專利第5,604,231號和Cosford等人的美國專利第5,852,041號,關于這些治療教導,通過引用將它們的內容并入本說明書。化合物及其藥物組合物可用于治療或預防多種CNS障礙,包括神經變性障礙、神經精神病學障礙、神經學障礙和成癮。化合物及其藥物組合物可用于治療或預防年齡相關性及其它認知缺陷和功能障礙;注意性障礙和癡呆,包括由感染因子或代謝紊亂所引起的那些;提供神經保護;治療驚厥和多發性腦梗塞;治療心境障礙,強迫癥和成癮行為;提供痛覺喪失;控制炎癥,諸如由細胞因子和核因子κB所介導的炎癥;治療炎性障礙;提供疼痛緩解;治療代謝障礙諸如糖尿病或代謝綜合征;治療感染,作為用于治療細菌、真菌和病毒感染的抗感染藥。CNS障礙可使用本發明的化合物和藥物組合物進行治療或預防的障礙、疾病和病況有:年齡相關性記憶損傷(AAMI)、輕度認知損傷(MCI)、年齡相關性認知下降(ARCD)、早老性癡呆、早發性阿爾茨海默病、老年性癡呆、阿爾茨海默病型癡呆、阿爾茨海默病、無癡呆的認知損傷(CIND)、萊維小體癡呆、HIV癡呆、AIDS癡呆綜合征、血管性癡呆、唐氏(Down)綜合征、頭創傷、創傷性腦損傷(TBI)、拳擊運動員癡呆、克-雅(Creutzfeld-Jacob)病和朊病毒疾病、卒中、缺血、注意缺陷障礙、注意渙散多動癥、誦讀困難、精神分裂癥、精神分裂癥樣障礙、分裂情感性障礙、精神分裂癥中的認知功能障礙、精神分裂癥中的認知缺陷諸如記憶(包括工作記憶)、執行功能、注意力、警惕性、信息處理和學習、與精神分裂癥有關的癡呆(無論是輕度、中度還是重度癡呆)、與精神分裂癥有關的癡呆(無論是輕度、中度還是重度癡呆)、帕金森神經功能障礙、包括帕金森病、腦炎后帕金森神經功能障礙、關島(Gaum)帕金森神經功能障礙-癡呆、帕金森病型額顳葉癡呆(FTDP)、皮克(Pick)病、尼-皮(Niemann-Pick)病、亨廷頓病、亨廷頓舞蹈病、遲發性運動障礙、運動機能亢進、進行性核上性麻痹、進行性核上性局部麻痹、不寧腿綜合征、多發性硬化、肌萎縮性側索硬化(ALS)、運動神經元疾病(MND)、多系統萎縮癥(MSA)、皮質基底節變性、格林-巴利(Guillain-Barre)綜合征(GBS)、慢性炎性脫髓鞘性多神經病(CIDP)、癲癇癥、常染色體顯性遺傳性夜間額葉癲癇、躁狂癥、焦慮癥、抑郁癥、月經前煩躁不安、驚恐癥、貪食癥、厭食癥、發作性睡病、日間過度嗜睡、雙相性精神障礙、泛代性焦慮癥、強迫觀念與行為障礙、暴怒、對立違抗障礙、圖雷特(Tourette)綜合征、孤獨癥、藥物和酒精成癮、煙草成癮、肥胖癥、惡病質、銀屑病、狼瘡、急性膽管炎、口瘡性口炎、潰瘍、哮喘、潰瘍性結腸炎、炎性腸病、局限性回腸炎、術后腸梗阻、痙攣性張力障礙、腹瀉、便秘、隱窩炎、胰腺炎、病毒性肺炎、關節炎、包括類風濕性關節炎和骨關節炎、內毒素血癥、膿毒病、動脈粥樣硬化、特發性肺纖維化、急性疼痛、慢性疼痛、神經病變、尿失禁、糖尿病和瘤形成。認知損傷或功能障礙可能與諸如以下的精神疾病或病況有關:精神分裂癥和其它精神病,包括但不限于,精神病,精神分裂癥樣障礙,分裂情感性障礙,妄想性障礙,短暫性精神病,共有型精神病和由一種或多種一般醫學病況所引起的精神病,癡呆,以及其它認知障礙,包括但不限于輕度認知損傷,早老性癡呆,阿爾茨海默病,老年性癡呆,阿爾茨海默病型癡呆,年齡相關性記憶損傷,萊維小體癡呆,血管性癡呆,AIDS癡呆綜合征,誦讀困難,包括帕金森病的帕金森神經功能障礙,帕金森病的認知損傷和癡呆,多發性硬化的認知損傷,由創傷性腦損傷所引起的認知損傷,由其它的一般醫學病況所引起的癡呆,焦慮癥,包括但不限于,無廣場恐怖癥的驚恐癥,伴有廣場恐怖癥的驚恐癥,無驚恐癥史的廣場恐怖癥,特異恐怖癥,社交恐怖癥,強迫觀念與行為障礙,創傷后精神緊張性障礙,急性精神緊張性障礙,泛化性焦慮癥和由一般醫學病況所引起的泛化性焦慮癥,心境障礙,包括但不限于重性抑郁障礙,心境惡劣性障礙,雙相性抑郁,雙相性躁狂,I型雙相性精神障礙,伴隨躁狂的抑郁癥,抑郁或混合性發作,II型雙相性精神障礙,循環性障礙和由一般醫學病況所引起的心境障礙,睡眠障礙,包括但不限于睡眠障礙,原發性失眠,原發性嗜睡,發作性睡病,深眠障礙,惡夢障礙,睡眠驚恐障礙和夢游障礙,腦力發育遲緩,學習障礙,運動技能障礙,交流障礙,廣泛性發育障礙,注意缺陷和分裂行為障礙,注意缺陷障礙,注意渙散多動癥,嬰兒、兒童或成人的進食和攝食障礙,痙攣障礙,排泄障礙,物質相關障礙,包括但不限于物質依賴,物質濫用,物質中毒,物質戒除,酒精相關障礙,安非他明或安非他明樣相關障礙,咖啡因相關障礙,大麻相關障礙,可卡因相關障礙,迷幻劑相關障礙,吸入劑相關障礙,煙堿相關障礙,鴉片樣物質相關障礙,苯環利定或苯環利定樣相關障礙,以及鎮靜藥、催眠要或抗焦慮藥相關障礙,人格障礙,包括但不限于,強迫觀念與行為人格障礙和沖動-控制障礙。精神分裂癥的癥狀一般被分為三類:陽性,陰性和認知癥狀。陽性癥狀,有時可被稱為“精神病性”癥狀,并包括錯覺和幻覺。“陽性”是指具有明顯的癥狀。陰性癥狀包括情緒平淡或缺乏表達,不能開始或完成活動,言語簡短和缺少內容,以及缺乏活動的愉快感或不興趣。“陰性”是指缺乏其它在健康受試者中存在的某些特征。認知癥狀屬于思維過程。認知癥狀包括認知缺陷,諸如包括工作記憶的記憶,執行功能,注意力,警惕性,信息處理和學習,參見Sharma等人的CognitiveFunctioninSchizophrenia:Deficits,FunctionalConsequences,andFutureTreatment,Psychiatr.Clin.N.Am.26(2003)25-40,在此通過引用將其并入本說明書。精神分裂癥還影響情緒。盡管許多罹患精神分裂癥的受試者變得抑郁,但是一些受試者具有明顯的情緒波動甚至具有雙相樣狀態。上述的病況和障礙在例如以下文獻中進一步被詳細討論:theAmericanPsychiatricAssociation:DiagnosticandStatisticalManualofMentalDisorders,第4版,TextRevision,Washington,DC,AmericanPsychiatricAssociation,2000;關于定義這些病況和障礙,通過引用將該參考文獻并入本說明書。還可參見該手冊關于與物質使用、濫用和依賴有關的癥狀和診斷特征的詳細描述。優選地,進行疾病、障礙和病況的治療或預防,而無明顯的不良副作用,包括例如血壓和心率的顯著升高,對胃腸道的顯著的不利影響,以及對骨骼肌的顯著影響。本發明的化合物,當以有效量被使用時,被認為調節α7NNR的活性,而與人神經節所特有的煙堿樣亞型無明顯的相互作用(通過在腎上腺嗜鉻組織內缺乏引發煙堿樣功能的能力來證明)或與骨骼肌所特有的煙堿樣亞型無明顯的相互作用(通過在表達肌肉型煙堿樣受體的細胞制備物中內缺乏引發煙堿樣功能的能力來證明)。因此,這些化合物被認為能夠治療或預防疾病、障礙和病況,而不引發與在神經節和神經肌肉部位處的活性有關的顯著的副作用。因此,化合物的給藥被認為提供了治療窗,在該治療窗內,提供了對某些疾病、障礙和病況的治療,并且避免了某些副作用。也就是說,有效劑量的化合物被認為足以為障礙、障礙或病況提供所需的效果,但是認為不足以(即,不在足夠高的水平下)提供不希望的副作用。因此,本發明提供了本發明的化合物或其藥學上可接受的鹽在治療中,諸如上述治療中的應用。在本發明的又一個方面,提供了本發明的化合物或其藥學上可接受的鹽在制備用于治療CNS障礙諸如上文所述的障礙、疾病或病況的藥物中的用途。炎癥已知神經系統(主要通過迷走神經)通過抑制巨噬細胞腫瘤壞死因子(TNF)的釋放來調節先天免疫應答的量級。這一生理機制被稱為“膽堿能抗炎途徑”(例如,參見Tracey,“Theinflammatoryreflex,”Nature420:853-9(2002),通過引用將其并入本說明書)。過量的炎癥因子和腫瘤壞死因子合成導致各種疾病的發病,甚至是死亡。這些疾病包括但不限于內毒素血癥,類風濕性關節炎,骨關節炎,銀屑病,哮喘,動脈粥樣硬化,特發性肺纖維化和炎性腸病。可通過給予本說明書所述的化合物來進行治療或預防的炎性病況包括但不限于:慢性和急性炎癥,銀屑病,內毒素血癥,痛風,急性假性痛風,急性痛風性關節炎,關節炎,類風濕性關節炎,骨關節炎,多肌炎,皮膚肌炎,強直性脊椎炎,斯提耳(Still)病,成年發病性斯提耳病,同種異體移植排斥,慢性移植排斥,哮喘,動脈粥樣硬化,單核細胞-巨噬細胞依賴性肺損傷,特發性肺纖維化,特應性皮炎,慢性阻塞性肺病,成人呼吸窘迫綜合征,在鐮狀細胞病中的急性胸綜合征,炎性腸病,局限性回腸炎,潰瘍性結腸炎,急性膽管炎,口瘡性口炎,隱窩炎,腎小球腎炎,狼瘡腎炎,血栓形成和移植物抗宿主反應。與細菌和/或病毒感染有關的炎性應答許多的細菌和/或病毒感染(例如,腦膜炎、肝炎和腎炎)與通過毒素的形成、身體對細菌或病毒和/或毒素的自然反應所引起的副作用有關。如上所述,身體對感染的反應通常牽涉產生大量的TNF和/或其它細胞因子。這些細胞因子的過量表達可導致嚴重損傷,諸如膿毒性休克(當細菌是膿毒病時),內毒素性休克,尿膿毒病和中毒性休克綜合征。細胞因子表達由NNR介導,并且可通過給予這些受體的激動劑或部分激動劑而受到抑制。因此,作為這些受體的激動劑或部分激動劑的本說明書所述的這些化合物可用于使得與細菌感染以及與病毒和真菌感染有關的炎性應答最小化。這些細菌感染的實例包括炭疽、肉毒中毒和膿毒病。這些化合物中的一些還具有抗微生物性質。這些化合物還可作為與目前用于處置細菌、病毒和真菌感染的治療劑諸如抗生素、抗病毒藥和抗真菌藥相組合的輔助治療劑。抗毒素也可用于結合至由感染因子所產生的毒素并使得被結合的毒素穿行通過身體而不產生炎性應答。抗毒素的實例被公開在例如Bundle等人的美國專利第6,310,043號中,通過引用將其并入本說明書。其它有效對抗細菌和其它毒素的藥劑可以是有效的并且它們的治療效果可通過與本說明書所述的化合物進行共同給藥而得以互補。疼痛化合物可被給藥用于治療和/或預防疼痛,包括急性疼痛、神經學疼痛、炎性疼痛、神經性疼痛和慢性疼痛。本說明書所述的化合物的止痛活性可以在持續性炎性疼痛和神經性疼痛模型中得以證明,這些模型如美國專利申請公開文本第20010056084A1號(Allgeier等人)中所述進行,通過引用將其并入本說明書,其中在炎性疼痛的完全弗氏(Freund)佐劑大鼠模型中表現痛覺過敏和在神經性疼痛的小鼠部分坐骨神經結扎模型中表現機械痛覺過敏。止痛效果適用于治療由各種原因或病因所導致的疼痛,特別用于治療炎性疼痛和有關的痛覺過敏,神經性疼痛和有關的痛覺過敏,慢性疼痛(例如,嚴重慢性疼痛,手術后疼痛,以及與包括癌、絞痛、腎或膽絞痛、月經、偏頭痛和痛風在內的各種病況有關的疼痛)。炎性疼痛可由不同的原因引起,包括關節炎和類風濕性病,腱滑膜炎和脈管炎。神經性疼痛包括三叉神經性神經痛或皰疹性神經痛,糖尿病性神經病性疼痛,灼性神經痛,下背痛,以及傳入神經阻滯綜合征諸如臂叢撕脫。新血管形成α7NNR與新血管形成有關。新血管形成的抑制,例如,通過給予α7NNR的拮抗劑(或在某些劑量下的部分激動劑),可以治療或預防以不希望的新血管形成或血管發生為特征的病況。這些病況可包括以炎性血管發生和/或由缺血誘導的血管發生為特征的那些。與瘤生長有關的新血管形成也可通過給予本說明書所述的起到α7NNR的拮抗劑或部分激動劑的作用的化合物得以抑制。α7NNR特異性活性的特異性拮抗減少對炎癥、缺血和瘤形成的血管發生應答。關于用于評價本說明書所述化合物的合適的動物模型系統的指導,可參見例如Heeschen,C.等人的"Anovelangiogenicpathwaymediatedbynon-neuronalnicotinicacetylcholinereceptors,"J.Clin.Invest.110(4):527-36(2002),關于血管發生的α7-特異抑制和與人類疾病有關的血管發生活性的細胞(體外)和動物模建,特別是關于Lewis肺腫瘤模型(體內,在小鼠中,特別參見第529和532-533頁),通過引用將其并入本說明書。可使用本說明書所述的化合物進行治療的代表性的腫瘤類型包括NSCLC,卵巢癌,胰腺癌,乳癌,結腸癌,直腸癌,肺癌,口咽癌,下咽癌,食管癌,胃癌,胰腺癌,肝癌,膽囊癌,膽管癌,小腸癌,尿道癌,腎癌,膀胱癌,泌尿道上皮癌,女性生殖道癌,子宮頸癌,子宮癌,卵巢癌,絨膜癌,妊娠期滋養層疾病,男性生殖道癌,前列腺癌,精囊腺癌,睪丸癌,生殖細胞瘤,內分泌腺癌,甲狀腺癌,腎上腺癌,腦下垂體癌,皮膚癌,血管瘤,黑素瘤,肉瘤,骨和軟組織肉瘤,卡波西肉瘤,腦瘤,神經瘤,眼瘤,腦膜瘤,星形細胞瘤,神經膠質瘤,成膠質細胞瘤,視網膜母細胞瘤,神經瘤,神經母細胞瘤,神經鞘瘤,腦膜瘤,由造血惡性病引起的實體瘤(諸如白血病,綠色瘤,漿細胞瘤,以及蕈樣真菌病的斑塊和腫瘤,以及皮膚T細胞淋巴瘤/白血病),以及得自淋巴瘤的實體瘤。所述化合物還可與其它形式的抗癌療法組合使用,包括與抗腫瘤藥諸如順鉑、阿霉素、柔紅霉素等共同給藥,以及/或與抗-VEGF(血管內皮細胞生長因子)藥劑共同給藥,這些是本領域已知的。化合物可以使它們靶向于腫瘤部位的方式被給藥。例如,化合物可以在與各種抗體綴合的微球、微粒或脂質體內被給藥,所述抗體引導所述的微粒子到達腫瘤。另外,化合物可以存在于這樣的微球、微粒或脂質體內,這些微球、微粒或脂質體經過適當地篩分以穿行通過動脈和靜脈、但進入并固定在腫瘤周圍的毛細管床內,并將所述化合物局部給予到腫瘤。這些藥物遞送裝置是本領域已知的。其它障礙除了治療CNS障礙、炎癥、新血管形成和疼痛之外,本發明的化合物還可用于預防或治療某些其它的其中NNR起作用的病況、疾病和障礙。實例包括自身免疫障礙諸如狼瘡,與細胞因子釋放有關的障礙,繼發于感染的惡病質(例如,在AIDS、AIDS相關綜合征和瘤形成中存在的),代謝障礙,包括I型糖尿病,II型糖尿病,代謝綜合征,肥胖癥或高血糖癥,pemphitis,尿失禁,視網膜疾病,感染性疾病,肌無力,類重癥肌無力(Eaton-Lambert)綜合征,高血壓,骨質疏松癥,血管收縮,血管舒張,心律不齊,貪食癥,厭食癥以及在公開的PCT申請WO98/25619中所述的那些適應癥,關于這些障礙,將該文獻并入本說明書。本發明的化合物還可被給予以治療驚厥,諸如作為癲癇癥狀的驚厥,并用于治療諸如梅毒和克-雅病等病況。診斷應用所述化合物可用于診斷用組合物,諸如探針,特別是當它們經過修飾以包括合適的標記物時。探針可用于例如確定特異性受體,特別是α7受體亞型的相對數目和/或功能。為此目的,本發明的化合物最優選用放射性同位素部分諸如11C、18F、76Br、123I或125I進行標記。被給予的化合物可采用適于所用標記的已知的檢測方法進行檢測。檢測方法的實例包括正電子發射斷層攝影術(PET)和單光子發射計算機斷層攝影術(SPECT)。如上所述的放射性標記物可用于PET(例如,11C,18F或76Br)和SPECT(例如,123I)成像中,11C的半衰期為約20.4分鐘,18F的半衰期為約109分鐘,123I的半衰期為約13小時,以及76Br的半衰期為約16小時。希望高的比放射性以使得在非飽和濃度下的被選擇的受體亞型顯像。被給予的劑量典型地低于毒性劑量并提供強對比圖象。化合物預計能夠在無毒水平下被給予。以放射性標記成像領域技術人員已知的方式來確定劑量。例如,參見London等人的美國專利第5,969,144號,關于給予這種化合物,通過引用將其并入本說明書。所述化合物可以使用已知的技術被給予。例如,參見London等人的美國專利第5,969,144號,關于這種給藥,通過引用將其并入本說明書。化合物可在并入其它成分(諸如可用于配制診斷組合物的那些類型的成分)的制劑組合物中被給予。根據本發明可使用的化合物最優選以高純度的形式被使用。參見Elmalch等人的美國專利第5,853,696號,關于這種分析,通過引用將其并入本說明書。在化合物被給予到受試者(例如人受試者)后,在受試者內存在的化合物可以通過合適的技術進行成像和定量化,以便指示被選擇的NNR亞型的存在、量和功能。除了人之外,化合物也可被給予到動物,諸如小鼠、大鼠、狗和猴。SPECT和PET成像可使用任何合適的技術和裝置來進行。參見Villemagne等人,Arneric等人(編)的NeuronalNicotinicReceptors:PharmacologyandTherapeuticOpportunities,235-250(1998)和Elmalch等人的美國專利第5,853,696號中,關于代表性成像技術,通過引用將它們各自并入本說明書。放射性標記的化合物以高親和性結合于選擇性NNR亞型(例如α7),并優選表現出很小的與其它煙堿性膽堿能受體亞型(例如,與肌肉和神經節有關的那些受體亞型)的非特異性結合。就這點而論,所述化合物可用作用于受試者體內的煙堿性膽堿能受體亞型的非侵害性成像的試劑,特別地用在腦內用于與各種CNS疾病和障礙有關的診斷。在一個方面,診斷組合物可用于診斷受試者諸如人患者的疾病的方法中。所述方法涉及對患者給予經過可檢測標記的本說明書所述的化合物,并檢測該化合物與被選擇的NNR亞型(例如,α7受體亞型)的結合。使用診斷工具諸如PET和SPECT的本領域技術人員可以使用本說明書所述的放射性標記化合物來診斷各種病況和障礙,包括與中樞神經系統和自主神經系統的功能障礙有關的病況和障礙。這些障礙包括多種CNS疾病和障礙,包括阿爾茨海默病,帕金森病和精神分裂癥。可進行評價的這些和其它的代表性疾病和障礙包括在Bencherif等人的美國專利5,952,339中所述的那些,該文獻在此通過引用將其并入本說明書。在另一個方面,診斷組合物可用于監控受試者諸如人患者的選擇性煙堿樣受體亞型的方法中。所述方法涉及對患者給予經過可檢測標記的本說明書所述的化合物,并檢測該化合物與被選擇的煙堿樣受體亞型即α7受體亞型的結合。受體結合本發明的化合物在與NNR亞型、特別是α7受體亞型結合的化合物的結合試驗中用作參比配體。為此目的,本發明的化合物優選用放射性同位素部分諸如3H或14C進行標記。V.合成實施例提供了以下的合成實施例來說明本發明而不認為該實施例對本發明的范圍構成限制。在這些實施例中,所有的份數和百分數都以重量計,除非另作說明。所有的溶液是含水的,除非另作說明。實施例1:小規模合成(2S,3R)-N-(2-((3-吡啶基)甲基)-1-氮雜雙環[2.2.2]辛烷-3-基)苯并呋喃-2-甲酰胺(化合物A)及其對映體(2R,3S)-N-(2-((3-吡啶基)甲基)-1-氮雜雙環[2.2.2]辛烷-3-基)苯并呋喃-2-甲酰胺2-((3-吡啶基)亞甲基)-1-氮雜雙環[2.2.2]辛烷-3-酮將氫氧化鉀(56g,0.54摩爾)溶解在甲醇(420mL)中。加入3-奎寧環酮鹽酸鹽(75g,0.49摩爾)并將混合物在周圍環境溫度攪拌30分鐘。加入3-吡啶甲醛(58g,0.54摩爾)并將混合物在周圍環境溫度攪拌16小時。反應混合物在此期間變成黃色,有固體粘結在燒瓶壁上。從壁上刮下固體并使其破碎。在快速攪拌下,加入水(390mL)。當固體溶解時,將混合物在4℃冷卻過夜。通過過濾收集晶體,用水洗滌,并空氣干燥,得到80g的黃色固體。通過將濾液濃縮到其原來體積的約10%并在4℃冷卻過夜得到第二批(8g)產物。兩批具有足夠的用于進一步轉化的純度(88g,82%收率)。2-((3-吡啶基)甲基)-1-氮雜雙環[2.2.2]辛烷-3-酮將2-((3-吡啶基)亞甲基)-1-氮雜雙環[2.2.2]辛烷-3-酮(20g,93mmol)懸浮在甲醇(200mL)中并用46mL的6M鹽酸處理。加入10%的炭載鈀(1.6g)并將混合物在25psi氫氣下振搖16小時。將混合物過濾通過硅藻土,并通過旋轉蒸發除去濾液中的溶劑。得到粗制2-((3-吡啶基)甲基)-1-氮雜雙環[2.2.2]辛烷-3-酮鹽酸鹽,為白色膠狀物(20g),將其隨后用2M氫氧化鈉(50mL)和氯仿(50mL)處理并攪拌1小時。分離氯仿層,將水相用2M氫氧化鈉(~5mL,足夠使pH升高到10)和飽和的氯化鈉水溶液(25mL)處理。將該含水混合物用氯仿(3×10mL)提取,將合并的氯仿提取物干燥(無水硫酸鎂)并通過旋轉蒸發進行濃縮。將殘余物(18g)溶解在溫熱的醚(320mL)中并冷卻到4℃。濾出該白色固體,用少量冷乙醚洗滌并空氣干燥。將濾液濃縮到其原來體積的約10%并在4℃冷卻,得到第二批。合并收率為16g(79%)。3-氨基-2-((3-吡啶基)甲基)-1-氮雜雙環[2.2.2]辛烷在氮氣下,向攪拌的2-((3-吡啶基)甲基)-1-氮雜雙環[2.2.2]辛烷-3-酮(3.00g,13.9mmol)在無水甲醇(20mL)中的溶液中加入1M的氯化鋅在醚中的溶液(2.78mL,2.78mmol)。在周圍環境溫度攪拌30分鐘后,將該混合物用固體甲酸銨(10.4g,167mmol)處理。在周圍環境溫度攪拌另外1小時后,分份加入固體氰基硼氫化鈉(1.75g,27.8mmol)。然后將反應在周圍環境溫度攪拌過夜并通過加入水(~5mL)使反應終止。將經淬滅的反應在5M氫氧化鈉(10mL)和氯仿(20mL)之間分配。水層用氯仿(20mL)提取,并將合并的有機層干燥(硫酸鈉),過濾并濃縮。得到2.97g的黃色膠狀物。GCMS分析顯示該產物是順式胺和反式胺的1:9的混合物,并一起存在痕量的相應的醇(98%的總質量回收率)。(2R,3S)和(2S,3R)-3-氨基-2-((3-吡啶基)甲基)-1-氮雜雙環[2.2.2]辛烷將二-對甲苯酰基-D-酒石酸(5.33g,13.8mmol)加入到粗制3-氨基-2-((3-吡啶基)甲基)-1-氮雜雙環[2.2.2]辛烷(6.00g,27.6mmol的1:9的順式/反式混合物)在甲醇(20mL)中的溶液中。完全溶解后,通過旋轉蒸發將澄清溶液濃縮形成固體物質。將固體溶解在最小量的煮沸甲醇(~5mL)中。將溶液緩慢地冷卻,首先冷卻到周圍環境溫度(1小時),然后在5℃歷時約4小時,并最后在-5℃過夜。通過抽濾收集沉淀的鹽,并從5mL的甲醇重結晶。空氣干燥得到1.4g的白色固體,將其在氯仿(5mL)和2M氫氧化鈉(5mL)之間分配。氯仿層和水層的5mL氯仿提取物被合并,干燥(無水硫酸鈉)并濃縮,得到無色的油狀物(0.434g)。該游離堿的對映體純度通過將一部分轉化成其N-(叔丁氧羰基)-L-脯氨酰胺、然后使用LCMS分析非對映純度(98%)來測定。用2M氫氧化鈉使從最初結晶得到的母液呈堿性(~pH11)并用氯仿(10mL)提取兩次。將氯仿提取物干燥(無水硫酸鈉)并濃縮,得到油狀物。將該胺(3.00g,13.8mmol)溶解在甲醇(10mL)中并用二-對甲苯酰基-L-酒石酸(2.76g,6.90mmol)處理。將混合物溫熱以幫助溶解,然后緩慢地冷卻到-5℃,在-5℃將其保持過夜。通過抽濾收集沉淀物,從甲醇重結晶并干燥。得到1.05g的白色固體。將該鹽轉化成游離堿(收率=0.364g),并如上文關于另一個對映體所述,使用脯氨酰胺方法測定對映體純度(97%)。N-(2-((3-吡啶基)甲基)-1-氮雜雙環[2.2.2]辛烷-3-基)苯并呋喃-2-甲酰胺的反式對映體A將氯磷酸二苯酯(0.35mL,0.46g,1.7mmol)滴加到苯并呋喃-2-甲酸(0.28g,1.7mmol)和三乙胺(0.24mL,0.17g,1.7mmol)在無水二氯甲烷(5mL)中的溶液中。在周圍環境溫度攪拌30分鐘后,加入(2S,3R)-3-氨基-2-((3-吡啶基)甲基)-1-氮雜雙環[2.2.2]辛烷(0.337g,1.55mmol)(其衍生自二-對甲苯酰基-D-酒石酸鹽)和三乙胺(0.24mL,0.17g,1.7mmol)在無水二氯甲烷(5mL)中的溶液。將反應混合物在周圍環境溫度攪拌過夜,然后用10%氫氧化鈉(1mL)處理。分離雙相混合物,在Genevac離心蒸發器上濃縮有機層。將殘余物溶解在甲醇(6mL)中并在C18硅膠柱上,使用含0.05%三氟乙酸的乙腈/水梯度作為洗脫液進行HPLC純化。濃縮所選擇的級分,將得到的殘余物在氯仿和飽和碳酸氫鈉水溶液之間分配,并蒸發氯仿,得到0.310g(42%收率)的白色粉末(GCMS純度為95%)。1HNMR(300MHz,CDCl3)δ8.51(d,1H),8.34(dd,1H),7.66(d,1H),7.58(dt,1H),7.49(d,1H),7.44(s,1H),7.40(dd,1H),7.29(t,1H),7.13(dd,1H),6.63(d,1H),3.95(t,1H),3.08(m,1H),2.95(m,4H),2.78(m,2H),2.03(m,1H),1.72(m,3H),1.52(m,1H)。隨后通過手性色譜分析,確定該物質(反式對映體A)與絕對構型是2S,3R(通過X射線結晶學分析確定)的物質相同。N-(2-((3-吡啶基)甲基)-1-氮雜雙環[2.2.2]辛烷-3-基)苯并呋喃-2-甲酰胺的反式對映體B將氯磷酸二苯酯(96μL,124mg,0.46mmol)滴加到苯并呋喃-2-甲酸(75mg,0.46mmol)和三乙胺(64μL,46mg,0.46mmol)在無水二氯甲烷(1mL)中的溶液中。在周圍環境溫度攪拌45分鐘后,加入(2R,3S)-3-氨基-2-((3-吡啶基)甲基)-1-氮雜雙環[2.2.2]辛烷(0.10g,0.46mmol)(其衍生自二-對甲苯酰基-L-酒石酸鹽)和三乙胺(64μL,46mg,0.46mmol)在無水二氯甲烷(1mL)中的溶液中。將反應混合物在周圍環境溫度攪拌過夜,然后用10%氫氧化鈉(1mL)處理。分離雙相混合物,有機層和水層的氯仿提取物(2mL)通過旋轉蒸發進行濃縮。將殘余物溶解在甲醇中并在C18硅膠柱上,使用含有0.05%三氟乙酸的乙腈/水梯度作為洗脫液進行HPLC純化。濃縮所選擇的級分,將得到的殘余物在氯仿和飽和碳酸氫鈉水溶液之間分配,并蒸發氯仿,得到82.5mg(50%收率)的白色粉末。NMR譜與2S,3R異構體的NMR譜相同。因為該物質(反式對映體B)的直接前體與2S,3R化合物(反式對映體A)的直接前體是對映體關系,因此反式對映體B的絕對構型被假定為是2R,3S。實施例2:大規模合成(2S,3R)-N-(2-((3-吡啶基)甲基)-1-氮雜雙環[2.2.2]辛烷-3-基)苯并呋喃-2-甲酰胺和(2S,3R)-N-(2-((3-吡啶基)甲基)-1-氮雜雙環[2.2.2]辛-3-基)-1-苯并呋喃-2-甲酰胺對甲苯磺酸鹽2-((3-吡啶基)亞甲基)-1-氮雜雙環[2.2.2]辛烷-3-酮在氮氣氣氛下,將3-奎寧環酮鹽酸鹽(8.25kg,51.0mol)和甲醇(49.5L)加入到100L的玻璃反應燒瓶中,所述反應燒瓶裝備有機械攪拌器、測溫探頭和冷凝器。通過添粉漏斗將氫氧化鉀(5.55kg,99.0mol)在約30分鐘內加入,導致反應溫度從50℃升高到56℃。在約2小時內,將3-吡啶甲醛(4.80kg,44.9mol)加入到反應混合物中。將得到的混合物在20℃±5℃攪拌至少12小時,并通過薄層色譜法(TLC)監控反應。反應完成后,將反應混合物過濾通過燒結玻璃漏斗并將濾餅用甲醇(74.2L)洗滌。將濾液濃縮,轉移到反應燒瓶中,并加入水(66.0L)。將懸浮液攪拌至少30分鐘,過濾,并將濾餅用水(90.0L)洗滌直到洗液的pH為7-9。將固體在50℃±5℃下真空干燥至少12小時,得到8.58kg(89.3%)的2-((3-吡啶基)亞甲基)-1-氮雜雙環[2.2.2]辛烷-3-酮。(2S)-2-((3-吡啶基)甲基)-1-氮雜雙環[2.2.2]辛烷-3-酮二-對甲苯酰基-D-酒石酸鹽在惰性氣氛下,將2-((3-吡啶基)亞甲基)-1-氮雜雙環[2.2.2]辛烷-3-酮(5.40kg,25.2mol)和甲醇(40.5L)加入到72L反應容器中,該反應容器裝備有機械攪拌器、測溫探頭、低壓氣體調節系統和壓力計。將頂部空間充滿氮氣,并將混合物攪拌以獲得澄清的黃色溶液。向該燒瓶中加入10%的炭載鈀(50%濕度)(270g)。使用真空泵將反應器的氣氛排空,并用氫氣置換頂部空間達到10-20英寸的水壓力。排氣和用氫氣加壓再重復2次,使得在第三次加壓后反應器處于20英寸的水壓力下的氫氣中,將反應混合物在20℃±5℃攪拌至少12小時,并通過TLC監控反應。反應完成后,將懸浮液過濾通過在燒結玻璃漏斗上的545墊(1.9kg),并將濾餅用甲醇(10.1L)洗滌。將濾液濃縮得到半固體,在氮氣氣氛下將該半固體轉移到200L反應燒瓶中,該反應燒瓶裝備有機械攪拌器、冷凝器和測溫探頭。將該半固體溶解在乙醇(57.2L)中,并加入二-對甲苯酰基-D-酒石酸(DTTA)(9.74kg,25.2mol)。將攪拌的反應混合物在回流下加熱至少1小時,將反應保持另外的至少12小時并同時將反應冷卻到15℃到30℃。使用桌面過濾器將懸浮液過濾,將濾餅用乙醇(11.4L)洗滌。將產品在周圍環境溫度下真空干燥,得到11.6kg(76.2%收率,59.5%的純度因數)的(2S)-2-((3-吡啶基)甲基)-1-氮雜雙環[2.2.2]辛烷-3-酮二-對甲苯酰基-D-酒石酸鹽。(2S,3R)-3-氨基-2-((3-吡啶基)甲基)-1-氮雜雙環[2.2.2]辛烷二-對甲苯酰基-D-酒石酸鹽將水(46.25L)和碳酸氫鈉(4.35kg,51.8mol)加入到200L燒瓶中。完全溶解后,加入二氯甲烷(69.4L)。加入(2S)-2-((3-吡啶基)甲基)-1-氮雜雙環[2.2.2]辛烷-3-酮二-對甲苯酰基-D-酒石酸鹽(11.56kg,19.19mol),并將反應混合物攪拌2-10分鐘。使層分離歷時至少2分鐘(當必要時加入另外的水(20L)以分層)。除去有機相并用無水硫酸鈉干燥。向剩余的水相中加入二氯甲烷(34.7L),并將懸浮液攪拌2-10分鐘。使層分離,歷時2-10分鐘。再次,除去有機相并用無水硫酸鈉干燥。再次如上所述重復用二氯甲烷(34.7L)提取水相。每次提取的樣品用于手性HPLC分析。通過過濾除去硫酸鈉,并將濾液濃縮,得到(2S)-2-((3-吡啶基)甲基)-1-氮雜雙環[2.2.2]辛烷-3-酮(4.0kg),為固體。在氮氣氣氛下,將(2S)-2-((3-吡啶基)甲基)-1-氮雜雙環[2.2.2]辛烷-3-酮(3.8kg)轉移到潔凈的100L玻璃反應燒瓶中,該反應燒瓶裝備有機械攪拌器和測溫探頭。加入無水四氫呋喃(7.24L)和(+)-(R)-α-甲基芐基胺(2.55L,20.1mol)。將異丙醇鈦(IV)(6.47L,21.8mol)在1小時內加入到攪拌的反應混合物中。將反應在氮氣氣氛下攪拌至少12小時。將乙醇(36.17L)加入到反應混合物中。將反應混合物冷卻到低于-5℃,并分份加入硼氫化鈉(1.53kg,40.5mol),保持反應溫度低于15℃(這一加料需要幾個小時)。然后將反應混合物在15℃±10℃攪拌至少1小時。通過HPLC監控反應,反應完成(由剩余低于0.5%的(2S)-2-((3-吡啶基)甲基)-1-氮雜雙環[2.2.2]辛烷-3-酮所顯示)后,加入2M氫氧化鈉(15.99L)并將混合物攪拌至少10分鐘。將反應混合物過濾通過桌面漏斗內的545墊。將濾餅用乙醇(15.23L)洗滌,并將濾液濃縮,得到油狀物。在惰性氣氛下將濃縮物轉移到潔凈的100L玻璃反應燒瓶中,該反應燒瓶裝備有機械攪拌器和測溫探頭。加入水(1L),并將混合物冷卻到0℃±5℃。將2M鹽酸(24L)加入到混合物中以調節混合物的pH到pH1。然后將混合物攪拌至少10分鐘,并緩慢地加入2M氫氧化鈉(24L)以調節混合物的pH到pH14。將混合物攪拌至少10分鐘,將水相用二氯甲烷(3×15.23L)提取。將有機相用無水硫酸鈉(2.0kg)干燥,過濾并濃縮,得到(2S,3R)-N-((1R)-苯基乙基)-3-氨基-2-((3-吡啶基)甲基))-1-氮雜雙環[2.2.2]辛烷(4.80kg,84.7%收率)。在惰性氣氛下,將(2S,3R)-N-((1R)-苯基乙基)-3-氨基-2-((3-吡啶基)甲基)-1-氮雜雙環[2.2.2]辛烷轉移到22L的玻璃燒瓶中,該燒瓶裝備有機械攪拌器和測溫探頭。加入水(4.8L),并將攪拌的混合物冷卻到5℃±5℃。將濃鹽酸(2.97L)緩慢地加入到反應燒瓶中,保持混合物的溫度低于25℃。在惰性氣氛下,將得到的溶液轉移到含有乙醇(18L)并裝備有機械攪拌器、測溫探頭和冷凝器的72L反應燒瓶中。向該燒瓶中加入10%的炭載鈀(50%濕度)(311.1g)和環己烯(14.36L)。將反應混合物在接近回流的溫度加熱至少12小時,并通過TLC監控反應。反應完成后,將反應混合物冷卻到低于45℃,將其過濾通過在燒結玻璃漏斗上的545墊(1.2kg)。將濾餅用乙醇(3L)漂洗并將濾液濃縮,得到水相。將水(500mL)加入到經過濃縮的濾液中,并將該合并的水層用甲基叔丁基醚(MTBE)(2x4.79L)洗滌。將2M氫氧化鈉(19.5L)加入到水相中以調節混合物的pH到pH14。然后將混合物攪拌至少10分鐘。將水相用氯仿(4×11.96L)提取,將合并的有機相用無水硫酸鈉(2.34kg)干燥。將濾液過濾并濃縮,得到(2S,3R)-3-氨基-2-((3-吡啶基)甲基)-1-氮雜雙環[2.2.2]辛烷(3.49kg,大于定量收率),為油狀物。在惰性氣氛下,將(2S,3R)-3-氨基-2-((3-吡啶基)甲基)-1-氮雜雙環[2.2.2]辛烷轉移到潔凈的100L反應燒瓶中,該反應燒瓶裝備有機械攪拌器、冷凝器和測溫探頭。加入乙醇(38.4L)和二-對甲苯酰基-D-酒石酸(3.58kg,9.27mol)。將反應混合物在溫和回流下加熱至少1小時。然后將反應混合物攪拌至少12小時同時使其冷卻到15℃到30℃。將得到的懸浮液過濾,將濾餅用乙醇(5.76L)洗滌。在惰性氣氛下,將濾餅轉移到潔凈的100L玻璃反應燒瓶中,該反應燒瓶裝備有機械攪拌器、測溫探頭和冷凝器。加入9:1的乙醇/水溶液(30.7L),將得到的漿料在溫和回流下加熱至少1小時。然后將反應混合物攪拌至少12小時同時使其冷卻到15℃到30℃。將混合物過濾器并將濾餅用乙醇(5.76L)洗滌。收集產品并在50℃±5℃真空干燥至少12小時,得到5.63kg(58.1%收率)的(2S,3R)-3-氨基-2-((3-吡啶基)甲基)-1-氮雜雙環[2.2.2]辛烷二-對甲苯酰基-D-酒石酸鹽。(2S,3R)-N-(2-((3-吡啶基)甲基)-1-氮雜雙環[2.2.2]辛烷-3-基)苯并呋喃-2-甲酰胺在惰性氣氛下,將(2S,3R)-3-氨基-2-((3-吡啶基)甲基)-1-氮雜雙環[2.2.2]辛烷二-對甲苯酰基-D-酒石酸鹽(3.64kg,5.96mol)和10%的氯化鈉水溶液(14.4L,46.4mol)加入到裝備有機械攪拌器的72L的玻璃反應燒瓶中。將5M氫氧化鈉(5.09L)加入到攪拌的混合物中以調節混合物的pH到pH14。然后將混合物攪拌至少10分鐘。將含水溶液用氯仿(4x12.0L)提取,將合并的有機層用無水硫酸鈉(1.72kg)干燥。將合并的有機層過濾,將濾液濃縮,得到(2S,3R)-3-氨基-2-((3-吡啶基)甲基)-1-氮雜雙環[2.2.2]辛烷(1.27kg),為油狀物。在惰性氣氛下,將(2S,3R)-3-氨基-2-((3-吡啶基)甲基)-1-氮雜雙環[2.2.2]辛烷轉移到裝備有機械攪拌器的50L玻璃反應燒瓶中。將二氯甲烷(16.5L)、三乙胺(847mL,6.08mol)、苯并呋喃-2-甲酸(948g,5.85mol)和O-(苯并三唑-1-基)-N,N,N,1-四甲基銨六氟磷酸鹽(HBTU)(2.17kg,5.85mol)加入到反應混合物中。將混合物在周圍環境溫度攪拌至少4小時,并通過HPLC監控反應。反應完成后,將10%的碳酸鉀水溶液(12.7L,17.1mol)加入到反應混合物中并將混合物攪拌至少5分鐘。分層并將有機相用10%的鹽水(12.7L)洗滌,分層并將有機相冷卻到15℃±10℃。將3M鹽酸(8.0L)緩慢地加入到反應混合物中以調節混合物的pH到pH1。然后將混合物攪拌至少5分鐘。使層分離歷時至少5分鐘。使用桌面過濾器對固體過濾。將濾液分層,并將水相和得自漏斗的固體轉移到反應燒瓶中。將3M氫氧化鈉(9.0L)分份緩慢地加入到燒瓶中以調節混合物的pH到pH14。將水相用二氯甲烷(2×16.5L)提取。將合并的有機相用無水硫酸鈉(1.71kg)干燥。將混合物過濾,將濾液濃縮,得到(2S,3R)-N-(2-((3-吡啶基)甲基)-1-氮雜雙環[2.2.2]辛烷-3-基)苯并呋喃-2-甲酰胺(1.63kg,77.0%收率),為黃色固體。(2S,3R)-N-(2-((3-吡啶基)甲基)-1-氮雜雙環[2.2.2]辛-3-基]苯并呋喃-2-甲酰胺對甲苯磺酸鹽將(2S,3R)-N-(2-((3-吡啶基)甲基)-1-氮雜雙環[2.2.2]辛烷-3-基)苯并呋喃-2-甲酰胺(1.62kg,4.48mol)和二氯甲烷(8.60kg)加入到小口大玻璃瓶中。通過HPLC分析測定溶液中物質的重量/重量%。將溶液濃縮形成油狀物,加入丙酮(4L),將混合物濃縮形成含油固體。加入另外的丙酮(12L)到位于旋轉蒸發器蒸發球中的含油固體中,在惰性氣氛下將得到的漿料轉移到裝備有機械攪拌器、冷凝器、測溫探頭和冷凝器的50L玻璃反應燒瓶中。將反應混合物加熱到50℃±5℃。將水(80.7g)加入溶液,并將其攪拌至少10分鐘。將對甲苯磺酸(853g,4.44mol)在約15分鐘內分份加入到反應混合物中。將反應混合物加熱到回流并在該溫度保持至少30分鐘以獲得溶液。將反應在約2小時內冷卻到40℃±5℃。在約1.5小時內加入乙酸異丙酯(14.1L)。在至少10小時內將反應混合物緩慢地冷卻到周圍環境溫度。將混合物過濾并將濾餅用乙酸異丙酯(3.5L)洗滌。將分離的產品在105℃±5℃真空干燥2-9小時,得到2.19kg(88.5%收率)的(2S,3R)-N-(2-((3-吡啶基)甲基)-1-氮雜雙環[2.2.2]辛-3-基)苯并呋喃-2-甲酰胺對甲苯磺酸鹽,mp226-228℃。1HNMR(500MHz,D2O)δ8.29(s,1H),7.78(m,J=5.1,1H),7.63(d,J=7.9,1H),7.54(d,J=7.8,1H),7.49(d,J=8.1,2H),7.37(m,J=8.3,1H),7.33(m,J=8.3,6.9,1.0,1H),7.18(m,J=7.8,6.9,1.0,1H),7.14(d,J=8.1,2H),7.09(s,1H),6.99(dd,J=7.9,5.1,1H),4.05(m,J=7.7,1H),3.74(m,1H),3.47(m,2H),3.28(m,1H),3.22(m,1H),3.15(dd,J=13.2,4.7,1H),3.02(dd,J=13.2,11.5,1H),2.19(s,3H),2.02(m,2H),1.93(m,2H),1.79(m,1H).13CNMR(126MHz,D2O)δ157.2,154.1,150.1,148.2,146.4,145.2,138.0,137.0,130.9,128.2(2),126.9,126.8,125.5(2),123.7,123.3,122.7,111.7,100.7,61.3,50.2,48.0,40.9,33.1,26.9,21.5,20.8,17.0。通過用氫氧化鈉水溶液處理并用氯仿提取,將該物質的樣品轉化為化合物A游離堿(用于鹽選擇研究)。充分蒸發氯仿產生灰白色粉末,mp167-170℃,該粉末具有以下的譜圖特征:正離子電噴射MS[M+H]+離子m/z=362。1HNMR(500MHz,DMSO-d6)δ8.53(d,J=7.6Hz,1H),8.43(d,J=1.7Hz,1H),8.28(dd,J=1.6,4.7Hz,1H),7.77(d,J=7.7Hz,1H),7.66(d,J=8.5Hz,1H),7.63(dt,J=1.7,7.7Hz,1H),7.52(s,1H),7.46(m,J=8.5,7.5Hz,1H),7.33(m,J=7.7,7.5Hz,1H),7.21(dd,J=4.7,7.7Hz,1H),3.71(m,J=7.6Hz,1H),3.11(m,1H),3.02(m,1H),2.80(m,2H),2.69(m,2H),2.55(m,1H),1.80(m,1H),1.77(m,1H),1.62(m,1H),1.56(m,1H),1.26(m,1H).13CNMR(126MHz,DMSO-d6)δ158.1,154.1,150.1,149.1,146.8,136.4,135.4,127.1,126.7,123.6,122.9,122.6,111.8,109.3,61.9,53.4,49.9,40.3,35.0,28.1,26.1,19.6。化合物A的一鹽酸鹽(參見實施例5)用于X射線結晶學分析。得到的晶體結構(分別如圖10A和10B所示)確認了化合物A的2S,3R絕對構型。實施例3:(2S,3R)-N-(2-((3-吡啶基)甲基)-1-氮雜雙環[2.2.2]辛-3-基)苯并呋喃-2-甲酰胺磷酸鹽的合成向圓底燒瓶中加入(2S,3R)-N-(2-((3-吡啶基)甲基)-1-氮雜雙環[2.2.2]辛-3-基)苯并呋喃-2-甲酰胺(8.18g,22.6mmol)和2-丙醇(180mL)。將混合物在65-70℃攪拌和加熱,直到所有固體溶解。將溶液在65-70℃劇烈攪拌,通過移液管滴加磷酸(1.65mL,24.3mmol)。立刻形成白色的粒狀固體。將混合物在65-70℃攪拌30分鐘,冷卻到周圍環境溫度(23℃)并攪拌另外24小時。通過吸濾收集白色固體,將濾餅用2-丙醇(20mL)洗滌并將固體空氣干燥至少1小時。將固體在真空箱中在75℃進一步干燥過夜(16小時),得到10.7g的產物(大于定量收率),熔點265-273℃(分解),在約180℃觀察到結晶度改變。1H-NMR(DMSO-d6)顯示存在2-丙醇(強溶劑合物),其可解釋大于定量收率的原因。手性LC分析得出97.1%的純度(270nm)。實施例4:(2S,3R)-N-(2-((3-吡啶基)甲基)-1-氮雜雙環[2.2.2]辛-3-基)苯并呋喃-2-甲酰胺馬來酸鹽的合成將馬來酸(0.067g,0.630mmol)加入到(2S,3R)-N-(2-((3-吡啶基)甲基)-1-氮雜雙環[2.2.2]辛-3-基)苯并呋喃-2-甲酰胺(0.203g,0.560mmol)在乙酸異丙酯(2mL)中的熱漿料中,有細的白色固體沉淀析出,同時還有膠質殘余物。加入另外的乙酸異丙酯(3mL)和馬來酸(0.006g),將混合物加熱至回流,在回流下加入異丙醇(5mL)。將得到的白色固體混合物冷卻到周圍環境溫度,過濾,將固體用乙酸異丙酯(2mL)洗滌。將產物在60℃真空干燥18小時,得到0.228g的灰白色的片狀固體(84.7%收率),mp180-182℃。1HNMR(DMSO-d6)顯示是單鹽化學計量。C22H23N3O2C4H4O4的計算值:C,65.40;H,5.70;N,8.80;實測值:C,65.35,65.29;H,5.86,5.68;N,8.69,8.78。實施例5:(2S,3R)-N-(2-((3-吡啶基)甲基)-1-氮雜雙環[2.2.2]辛-3-基)苯并呋喃-2-甲酰胺鹽酸鹽的合成一鹽酸鹽:通過將濃鹽酸(1.93mL,12M,23.2mmol)滴加到8.5mL的冷THF中來制備鹽酸/THF溶液。將該溶液溫熱到周圍環境溫度。向圓底燒瓶中加入(2S,3R)-N-(2-((3-吡啶基)甲基)-1-氮雜雙環[2.2.2]辛-3-基)苯并呋喃-2-甲酰胺(8.49g,23.5mmol)和丙酮(85mL)。將混合物在45-50℃攪拌和加熱,直到實現完全溶解。在5分鐘內滴加上面制備的鹽酸/THF溶液,使用另外的THF(1.5mL)用于轉移。在加入酸性溶液期間開始形成粒狀的白色固體。將混合物冷卻到周圍環境溫度,并攪拌過夜(16小時)。通過抽濾收集固體,將濾餅用丙酮(10mL)洗滌,將固體在抽吸下空氣干燥30分鐘。將固體在真空箱中在75℃另外干燥2小時,得到8.79g的細的白色晶體(94%收率),mp255-262℃。手性LC分析得到98.8%的純度(270nm)。1H-NMR(DMSO-d6)顯示無殘留溶劑并證實單化學計量。1HNMR(300MHz,DMSO-d6)δ10.7(寬的s,1H-季銨),8.80(寬的s,1H-酰胺H),8.54(s,1H),8.23(d,1H),7.78(d,1H),7.74(d,1H),7.60(d,1H),7.47(m,2H),7.33(m,1H),7.19(m,1H),4.19(m,1H),4.08(m,1H),3.05-3.55(m,6H),2.00-2.10(m,3H),1.90(m,1H),1.70(m,1H)。該鹽的X射線結晶學分析確認了立體化學歸屬和化學計量(參見圖10A和10B)。二鹽酸鹽:將氯化氫氣體緩慢地鼓泡通過(2S,3R)-N-(2-((3-吡啶基)甲基)-1-氮雜雙環[2.2.2]辛-3-基)苯并呋喃-2-甲酰胺(1.9g,5.3mmol)在無水乙醚(25mL)中的冰冷卻的溶液中。首先在氮氣氣流中,然后使用高真空(在高真空管中的氫氧化鈉洗滌器)除去揮發物。將殘余物與小體積的無水乙醚(被廢棄)研磨,并將剩余的固體高真空干燥。得到2.17g(94%收率)灰白色粉末,mp210-212℃(吸濕的)。手性LC分析得出93.7%的純度(270nm)。正離子電噴射MS[M+H]+離子m/z=362。1HNMR(300MHz,CD3OD)δ9.15(s,1H),8.84(d,1H),8.63(d,1H),7.97(t,1H),7.75(d,1H),7.61(d,1H),7.52(m,2H),7.35(t,1H),4.50(m,1H),4.32(m,1H),3.40-3.85(m,6H),1.95-2.40(m,5H)。實施例6:(2S,3R)-N-(2-((3-吡啶基)甲基)-1-氮雜雙環[2.2.2]辛-3-基)苯并呋喃-2-甲酰胺半半乳糖二酸鹽的合成將半乳糖二酸(粘酸)(36.3mg,0.173mmol)加入到(2S,3R)-N-(2-((3-吡啶基)甲基)-1-氮雜雙環[2.2.2]辛-3-基)苯并呋喃-2-甲酰胺(125mg,0.346mmol)在熱乙醇(1mL)中的溶液中。當加入水(8滴)時將混合物回流;然后使熱混合物過濾通過棉花塞,隨后將其用乙醇(1mL)漂洗。冷卻不能得到沉淀物。通過旋轉蒸發除去揮發物,將殘余物(白色泡沫)與異丙醇(被廢棄)研磨,將剩余的固體溶解在回流的丙酮/水(4mL,7:1)中。緩慢冷卻到5℃,產生白色固體,將其濾出,用異丙醇(3×1mL)洗滌,并高真空干燥。這樣得到118mg(73%收率)的細白色片狀物,mp134-139℃。1HNMR(300MHz,D2O)δ8.29(s,1H),7.78(d,1H),7.62(d,1H),7.54(d,1H),7.35(m,2H),7.18(t,1H),7.10(s,1H),6.98(m,1H),4.08(s,1H,半乳糖二酸),3.98(d,1H),3.77(s,1H,半乳糖二酸),3.66(m,1H),3.35(m,1H),2.95-3.30(m,4H),1.65-2.05(m,5H)。實施例7:(2S,3R)-N-(2-((3-吡啶基)甲基)-1-氮雜雙環[2.2.2]辛-3-基)苯并呋喃-2-甲酰胺D-酒石酸鹽的合成將酒石酸(25.6mg,0.173mmol)加入到(2S,3R)-N-(2-((3-吡啶基)甲基)-1-氮雜雙環[2.2.2]辛-3-基)苯并呋喃-2-甲酰胺(125mg,0.346mmol)在熱乙醇(1mL)中的溶液中。將得到的溶液緩慢地冷卻到周圍環境溫度。無固體沉淀析出,因此將溶液濃縮,得到白色泡沫。在異丙醇中結晶的嘗試以失敗告終。將該泡沫溶解在甲醇中并加入另外一半當量的酒石酸(25.6mg,0.173mmol)。將混合物濃縮,得到白色泡沫,其不從甲醇和異丙醇的混合物中結晶。然后將經過濃縮的物質(固體和膠狀液體的混合物)在乙酸乙酯(1mL)中成漿料,得到白色固體。其通過過濾進行分離(用乙酸乙酯洗滌)并在真空箱中干燥(18小時,在40℃),得到141mg(79.7%收率)的單化學計量的鹽(NMR),mp136-140℃。手性LC分析得到98.1%的純度(270nm)。1HNMR(300MHz,D2O)δ8.50(s,1H),8.01(d,1H),7.86(d,1H),7.75(d,1H),7.56(m,2H),7.38(t,1H),7.32(s,1H),7.21(t,1H),4.34(s,2H,酒石酸),4.26(d,1H),3.95(m,1H),3.64(m,2H),3.15-3.55(m,4H),1.90-2.30(m,5H)。實施例8:(2S,3R)-N-(2-((3-吡啶基)甲基)-1-氮雜雙環[2.2.2]辛-3-基)苯并呋喃-2-甲酰胺甲磺酸鹽的合成將甲磺酸(33.2mg,0.346mmol)加入到(2S,3R)-N-(2-((3-吡啶基)甲基)-1-氮雜雙環[2.2.2]辛-3-基)苯并呋喃-2-甲酰胺(125mg,0.346mmol)在熱乙醇(1mL)中的溶液中。冷卻不能產生沉淀物。將混合物回流,并使熱混合物過濾通過棉花塞,隨后將其用甲醇(1mL)漂洗。通過旋轉蒸發除去揮發物,并將殘余物(淺黃色泡沫)溶解在熱異丙醇(1mL)中。再次,冷卻不能產生沉淀物。蒸發異丙醇,并將殘余物在丙酮(1mL)中成漿料。過濾并在真空箱中干燥(18小時,在50℃),得到146mg(92.5%收率)的淺米色固體,mp240-243℃。1HNMR(300MHz,D2O)δ8.32(s,1H),7.82(d,1H),7.66(d,1H),7.57(d,1H),7.38(m,2H),7.20(m,1H),7.12(s,1H),7.01(m,1H),4.09(d,1H),3.75(m,1H),3.47(m,2H),3.00-3.40(m,4H),2.67(s,3H,甲磺酸),1.75-2.15(m,5H)。實施例9:(2S,3R)-N-(2-((3-吡啶基)甲基)-1-氮雜雙環[2.2.2]辛-3-基)苯并呋喃-2-甲酰胺D-扁桃酸鹽的合成將D-扁桃酸(52.6mg,0.346mmol)加入到(2S,3R)-N-(2-((3-吡啶基)甲基)-1-氮雜雙環[2.2.2]辛-3-基)苯并呋喃-2-甲酰胺(125mg,0.346mmol)在熱乙醇(1mL)中的溶液中。用乙酸乙酯(4ml)稀釋并冷卻不能產生沉淀物。通過旋轉蒸發除去揮發物,并將殘余物(白色泡沫)溶解在熱異丙醇(0.5mL)中。冷卻到5℃,產生白色結晶,通過抽濾收集該結晶。真空箱干燥(18小時,在45℃),得到111mg(62.4%收率)的淺米色固體,mp188.5-193℃。1HNMR(300MHz,D2O)δ8.33(s,1H),7.83(s,1H),7.67(d,1H),7.60(d,1H),7.27(m,8H,包含扁桃酸),7.12(s,1H),7.01(m,1H),4.85(s,1H,扁桃酸),4.10(d,1H),3.75(m,1H),3.48(m,2H),3.00-3.40(m,4H),1.75-2.15(m,5H)。實施例10:(2S,3R)-N-(2-((3-吡啶基)甲基)-1-氮雜雙環[2.2.2]辛-3-基)苯并呋喃-2-甲酰胺R-樟腦磺酸鹽的合成將R-10-樟腦磺酸(80.3mg,0.346mmol)加入到(2S,3R)-N-(2-((3-吡啶基)甲基)-1-氮雜雙環[2.2.2]辛-3-基)苯并呋喃-2-甲酰胺(125mg,0.346mmol)在熱乙醇(1mL)中的溶液中。冷卻不能沉積任何沉淀物。通過旋轉蒸發除去揮發物,并將殘余物(白色泡沫)溶解在熱異丙醇(0.5mL)中。冷卻到5℃產生很少的白色結晶和乳狀懸浮液。用刮鏟刮擦燒瓶側壁使混合物最終轉化成細白色結晶的厚塊。加入另外0.5mL的異丙醇,通過抽濾收集結晶。真空箱干燥(在70℃歷時5小時,然后在110℃歷時2小時),得到193mg(93.8%收率)的白色固體。mp149.5-156℃。1HNMR(300MHz,D2O)δ8.30(s,1H),7.79(d,1H),7.64(d,1H),7.55(d,1H),7.36(m,2H),7.18(m,1H),7.11(s,1H),6.99(m,1H),4.07(d,1H),3.73(m,1H),3.45(m,2H),3.95-3.35(m,5H,包含樟腦磺酸),2.64(d,1H,樟腦磺酸),2.22(m,2H),1.70-2.10(m,8H,包含樟腦磺酸),1.45(m,1H,樟腦磺酸),1.25(m,1H,樟腦磺酸),0.85(s,3H,樟腦磺酸),0.68(s,3H,樟腦磺酸)。實施例11:(2S,3R)-N-(2-((3-吡啶基)甲基)-1-氮雜雙環[2.2.2]辛-3-基)苯并呋喃-2-甲酰胺S-樟腦磺酸鹽的合成將S-10-樟腦磺酸(80.3mg,0.346mmol)加入到(2S,3R)-N-(2-((3-吡啶基)甲基)-1-氮雜雙環[2.2.2]辛-3-基)苯并呋喃-2-甲酰胺(125mg,0.346mmol)在熱乙醇(1mL)中的溶液。用乙酸乙酯(4mL)稀釋,冷卻不能沉積任何沉淀物。通過旋轉蒸發除去揮發物,并將殘余物(白色泡沫)溶解在熱異丙醇(1.5mL)中。冷卻到5℃,產生白色結晶。將混合物濃縮到約0.5mL并再次冷卻到5℃。然后通過抽濾收集固體并最初在45℃真空干燥18小時,然后在持續更高的溫度(最終達110℃)進行真空干燥以除去殘余的異丙醇。這得到143mg(69.7%收率)的白色固體,mp153.5-157℃。1HNMR(300MHz,D2O)δ8.29(s,1H),7.79(d,1H),7.63(d,1H),7.54(d,1H),7.34(m,2H),7.18(m,1H),7.10(s,1H),6.99(m,1H),4.05(d,1H),3.73(m,1H),3.44(m,2H),3.95-3.35(m,5H,包含樟腦磺酸),2.67(d,1H,樟腦磺酸),2.23(m,2H),1.70-2.10(m,8H,包含樟腦磺酸),1.46(m,1H,樟腦磺酸),1.25(m,1H,樟腦磺酸),0.84(s,3H,樟腦磺酸),0.64(s,3H,樟腦磺酸)。使用與上文所述的過程(實施例3-11)類似的過程,表征了幾種其它的鹽形式。這些制備物的結果在實施例12-14中記載。實施例12:(2S,3R)-N-(2-((3-吡啶基)甲基)-1-氮雜雙環[2.2.2]辛-3-基)苯并呋喃-2-甲酰胺硫酸鹽的合成硫酸鹽從乙酸異丙酯和水的混合物中沉淀析出。MP278℃。1HNMR(400MHz,DMSO-d6)δ9.28(寬的s,1H,酰胺),8.56(dd,1H),8.24(t,1H),7.77(d,1H),7.74(d,1H),7.60(s,1H),7.40(m,2H),7.35(s,1H),7.21(m,1H),4.21(m,1H),3.93(m,2H),3.10-3.60(m,5H),2.05(m,3H),1.92(m,1H),1.73(m,1H)。實施例13:(2S,3R)-N-(2-((3-吡啶基)甲基)-1-氮雜雙環[2.2.2]辛-3-基)苯并呋喃-2-甲酰胺酮戊二酸鹽的合成α-酮戊二酸鹽從乙酸異丙酯中沉淀析出。MP177℃。1HNMR(400MHz,DMSO-d6)δ8.64(s,1H,酰胺),8.50(d,1H),8.20(d,1H),7.74(d,1H),7.70(d,1H),7.60(m,1H),7.45(m,1H),7.32(m,2H),7.18(m,1H),4.10(m,1H),3.78(m,2H),3.00-3.45(m,5H),2.81(m,2H,酮戊二酸),2.41(m,2H,酮戊二酸),1.96(m,3H),1.83(m,1H),1.60(m,1H)。實施例14:(2S,3R)-N-(2-((3-吡啶基)甲基)-1-氮雜雙環[2.2.2]辛-3-基)苯并呋喃-2-甲酰胺馬尿酸鹽的合成馬尿酸鹽從丙酮中沉淀析出(吸濕性太強,以致于不能獲得熔點)。1HNMR(400MHz,DMSO-d6)δ8.79(s,1H,酰胺),8.56(d,1H),8.44(s,1H,馬尿酸),8.29(m,1H),7.87(m,2H,馬尿酸),7.76(d,1H),7.65(m,1H),7.54(m,1H),7.49(m,4H,包含馬尿酸),7.34(m,2H),7.21(m,1H),3.91(m,1H),3.74(m,2H),3.00-3.50(m,5H),2.80(m,2H,馬尿酸),1.79(m,2H),1.60(m,2H),1.30(m,1H)。實施例15:分離(2R,3R)-和(2S,3S)-N-(2-((3-吡啶基)甲基)-1-氮雜雙環[2.2.2]辛-3-基)苯并呋喃-2-甲酰胺和轉化成半乳糖二酸鹽通過旋轉蒸發,將從分離(2S,3R)-N-(2-((3-吡啶基)甲基)-1-氮雜雙環[2.2.2]辛-3-基]苯并呋喃-2-甲酰胺對甲苯磺酸鹽(實施例2)得到的上清液的樣品濃縮,用10%的氫氧化鈉水溶液調節到pH10并用二氯甲烷提取。蒸發二氯甲烷提取物,將殘余物(1.8g)溶解在含有0.5%二正丁胺的絕對乙醇(55mL)中。以每份0.25mL將該溶液注射到25cm×2.1cmAD手性HPLC柱上,用60:40:0.2的己烷/乙醇/二正丁胺(流速=30mL/min)洗脫,在270nm監控。分離在~7.5分鐘洗脫出的洗脫物和在~13.5分鐘洗脫出的洗脫物,在蒸發溶劑后分別得到0.48g(98%手性純度)和0.47g(99%手性純度)的無色油狀物。兩個NMR譜相同。1HNMR(300MHz,CDCl3)δ8.49(s,1H),8.45(d,1H),7.74(d,1H),7.52(m,4H),7.35(t,1H),7.20(dd,1H),7.05(d,1H),4.55(dt,1H),3.43(m,1H),3.22(m,1H),2.90(m,5H),2.09(m,1H),1.88(m,4H)。將每個游離堿樣品在無水乙醇(10mL)中的溫熱的溶液用一當量的半乳糖二酸處理。在攪拌下,將得到的混合物在75℃加熱5分鐘并冷卻到周圍環境溫度。抽濾收集得到的固體并真空干燥,分別得到0.65g(87%收率)和0.62g(85%收率)的白色粒狀固體(在各自的情況下,mp200-205℃)。1HNMR(300MHz,D2O)δ8.38(s,1H),8.28(d,1H),7.94(d,1H),7.70(d,1H),7.59(d,1H),7.48(t,1H),7.40(m,1H),7.32(m,2H),4.42(m,1H),4.21(s,2H),3.87(s,2H),3.68(m,1H),3.35(m,6H),2.25(m,2H),2.02(m,3H)。實施例16:(2R,3R)-N-(2-((3-吡啶基)甲基)-1-氮雜雙環[2.2.2]辛-3-基)苯并呋喃-2-甲酰胺對氯苯甲酸鹽的合成將固體對氯苯甲酸(46.8mg,0.299mmol)一次性加入到得自實施例15的先前洗脫的N-(2-((3-吡啶基)甲基)-1-氮雜雙環[2.2.2]辛-3-基]苯并呋喃-2-甲酰胺異構體(108mg,0.299mmol)在丙酮(10mL)中的溶液中。將該混合物升溫到接近回流,持續30分鐘并冷卻到周圍環境溫度。無沉淀物形成,因此將溶液濃縮到其原來體積的約20%(加熱板),此時開始形成結晶。將混合物冷卻并用異丙醇(2mL)稀釋。通過在周圍環境溫度緩慢蒸發溶劑將該混合物濃縮,收集得到的固體并干燥。這產生145mg(94%收率)的淺黃色結晶,mp150-152℃。1HNMR(300MHz,CDCl3)δ8.49(s,1H),8.38(d,1H),7.93(d,2H,對氯苯甲酸),7.67(m,2H),7.57(d,1H),7.45(m,1H),7.36(d,2H,對氯苯甲酸),7.30(m,1H),7.27(s,1H),7.16(m,1H),7.00(d,1H,酰胺),6.90(寬的s,季銨),4.62(m,1H),3.85(dd,1H),3.36(m,1H),2.95-3.25(m,5H),2.16(s,1H),1.70-2.10(m,4H)。該樣品的X射線結晶學分析揭示了其絕對立體化學是2R,3R(參見圖11A和11B)。因此通過排除法,在實施例15中的后一種被洗脫的異構體具有2S,3S的絕對構型。實施例17:用于立體異構體分析的手性色譜方法獲得用于將四個立體異構體彼此分離的手性色譜方法被證實非常具有挑戰性。最初的企圖(使用己烷/異丙醇/三乙胺流動相)導致峰重疊并且未達到最佳峰形。將異丙醇換成乙醇并將三乙胺換成二正丁胺,改善了分辨率和峰形并縮短了運行時間。該方法詳細描述如下:分析柱:AD(250×4.6mm,5μm)流動相:60:40:0.2的己烷/乙醇/二正丁胺注射體積:10μl流速:1.0毫升/分鐘溫度:20℃檢測:在270nm的UV總運行時間:~25分鐘洗脫順序(RT):2S,3R(5.3分鐘);2R,3S(7.3分鐘);2R,3R(8.3分鐘);2S,3S(12.1分鐘)立體異構體類似物的代表性色譜如圖12所示。實施例18:XRPD對本說明書描述的幾種鹽樣品進行XRPD分析。提供了鹽酸鹽(圖13)和甲苯磺酸鹽(圖14)的衍射圖樣。X射線粉末衍射(XRPD)收集了一個或兩個儀器的X射線粉末衍射圖樣。在SiemensD5000衍射儀上收集了一些圖樣,該SiemensD5000衍射儀使用CuKα輻射(40kV,40mA),θ-θ測角器,V20發散度和接收縫隙,石墨二級單色器和閃爍計數器。使用經證明的金剛砂標準品(NIST1976)進行儀器的性能檢查。使用原樣粉末在周圍環境條件下將運行樣品制成平板樣品。將約35mg的樣品輕輕地填塞進被切成經過研磨的零-背景(510)硅片的腔內。在分析期間將樣品在其自身平面內旋轉,以0.05°步長從2°到42°范圍掃描,每步4秒,使用CuKα1在BrukerAXSC2GADDS衍射儀上收集一些X射線粉末衍射圖樣,該BrukerAXSC2GADDS衍射儀使用CuKα輻射(40kV,40mA),自動化XYZ平臺,用于樣品自動定位的激光視頻顯微鏡和HiStar2-維區域檢測器。X射線光學系統包括與0.3mm的針孔準直儀連接的單一的多層鏡。光束發散度(即,樣品上X射線束的有效尺寸)是約4mm。使用θ-θ連續掃描方式,采用20cm的樣品-檢測器距離,這得到3.2°-30.0°的有效2θ范圍。典型地,樣品將暴露于X射線束下,歷時120秒。使用不經研磨的原樣粉末在周圍環境條件下將運行樣品制成平板樣品。將約1-2mg的樣品輕輕地按壓在硅片上以獲得扁平表面。在非周圍環境條件下將運行樣品設置在具有導熱化合物的硅片上。然后以大約10℃/分鐘將樣品加熱到合適的溫度并隨后等溫保持約5分鐘,然后開始收集數據。差示掃描量熱法(DSC)在裝備有50位自動進樣器的TA儀器Q1000上收集DSC數據。使用經證明的銦對儀器進行能量校準和溫度校準。典型地,將在針孔式鋁鍋中的0.5-1.5mg的每個樣品以10℃/分鐘從25℃加熱到175-200℃。在樣品上方保持30mL/min的氮氣吹掃。熱解重量分析法(TGA)在裝備有16位自動進樣器的TA儀器Q500TGA上收集TGA數據。使用經證明的Alumel對儀器進行溫度校準。典型地,將5-10mg的每個樣品裝載到預先稱皮重的鉑坩堝和鋁DSC鍋中,并以10℃/min從周圍環境溫度加熱到350℃。在樣品上方保持60mL/min的氮氣吹掃。偏振光顯微術(PLM)在具有用于捕捉圖像的數字攝像機的LeicaLM/DM偏振光顯微鏡上研究樣品。將少量的每個樣品置于載玻片上,將其置于浸油中并用蓋玻片覆蓋,盡可能使單個顆粒充分地分離。使用合適的放大倍數和連接至λ偽色過濾器的部分偏振光來觀察樣品。熱臺顯微術(HSM)使用與Mettler-ToledoMTFP82HT熱臺和用于捕捉圖像的數字攝像機組合的LeicaLM/DM偏振光顯微鏡進行熱臺顯微術。將少量的每個樣品置于載玻片上,使得單個顆粒盡可能充分地分開。使用合適的放大倍數和連接至λ偽光過濾器的部分偏振光來觀察樣品,同時以典型的10℃/分鐘從周圍環境溫度進行加熱。重量蒸氣吸附(GVS)在一個或兩個儀器上測定吸附等溫線。一些樣品使用受VTIFlowSystem4軟件控制的VTICorporationSGA-100水分吸附分析儀進行實驗。借助于Polyscience恒溫浴將樣品溫度保持在25℃。通過混合干和濕氮氣流控制濕度。使用精確度為+/-0.0001g的CahnDigitalRecordingBalanceD-200監控作為%RH的函數的重量改變。典型地,在周圍環境條件下將10-20mg的樣品置于稱皮重的天平盤中。將樣品在50℃干燥1小時。在25℃,以5%RH為間隔,在5-95%RH范圍內繪制標準品吸附等溫線,并且類似地,在25℃下,以5%RH為間隔,在95-5%的RH范圍內繪制解吸等溫線。對于每個%RH數據點,樣品平衡標準在5分鐘或180分鐘的最大平衡時間內包括0.0100wt%。使用受CFRSorp軟件控制的HidenIGASorp水分吸附分析儀獲得一些吸附等溫線。通過Huber再循環式水浴鍋將樣品溫度保持在25℃。通過將干和濕氮氣流混合來控制濕度,使用的總流速為250mL/min。通過位于樣品附近的經校準的VaisalaRH探針(動態范圍0-95%RH)測量相對濕度。通過測微天平(精確度±0.001mg)不間斷地監控樣品的相對于RH%的重量改變(質量減輕)。典型地,在周圍環境條件將10-20mg的樣品置于稱皮重的網格不銹鋼吊籃中。在40%RH和25℃(典型的周圍環境條件)下進行樣品的裝載和卸載。如下所述繪制水分吸附等溫線(2次掃描得出1個完整循環)。在25℃下,以10%RH為間隔,在0-90%的RH范圍內,繪制標準等溫線。GVS通用法參數參數值吸附-掃描140-90解吸/吸附-掃描285-干,干-40間隔(%RH)10掃描數2流速(mL/min)250溫度(℃)25穩定性(℃/min)0.05最小吸附時間(小時)1最大吸附時間(小時)4模式AF2精確度(%)98軟件使用最小二乘方最小化過程以及質量減輕模型,來預測漸近值。測量的質量減輕值必須處在軟件預測值的5%范圍內,然后選擇下一個RH%值。最小平衡時間被設為1小時,最大平衡時間被設為4小時。典型地,在完成等溫線后回收樣品并通過XRPD再次進行分析。通過KarlFischer法(KF)測量水在使用HydranalCoulomatAG試劑和氬吹掃的MettlerToledoDL39庫侖計上測量每個樣品的水含量。將稱重的固體樣品引入到容器內的鉑TGA盤上,該盤連接至subaseal以避免水進入。每次滴定使用大約10mg的樣品,并且進行一式兩份的測定。通過HPLC測定熱動力學水溶解度通過將足夠的化合物懸浮在0.25mL的水中以獲得最大的最終濃度為≥10mg/mL的無母體形式的化合物來測定水溶解度。將懸浮液在25℃平衡24小時,然后測量pH。然后將懸浮液過濾通過玻璃纖維C過濾器,進入96孔板。然后將濾液進行101倍稀釋。相對于約0.1mg/mL的在DMSO中的標準溶液進行HPLC定量。注射不同體積的標準品、經過稀釋的和未經稀釋的樣品溶液。使用峰面積計算溶解度,通過積分在與標準品注射中的主峰相同的保留時間發現的峰而測定所述峰面積。如果在過濾器板中存在足夠的固體,則收集XRPD。用于熱動力學水溶解度方法的通用法的詳細描述通過HPLC測定化學純度在裝備有二極管陣列檢測器并使用ChemStation軟件v9的AgilentHP1100串聯系統上進行純度分析。使用下文詳述的兩個方法之一。方法1方法2離子色譜法在使用ICNet軟件v2.3的Metrohm861AdvancedCompactIC上收集數據。將樣品制備成在水中的1000ppm母液。當樣品的溶解度低時,使用合適的共溶劑,諸如DMSO。在試驗之前,使用合適的溶劑將樣品稀釋到50ppm或100ppm。通過與已知濃度的正被分析的離子的標準溶液相比來進行定量測定。陰離子的離子色譜法陽離子的離子色譜法將約50mg的(2S,3R)-N-(2-((3-吡啶基)甲基)-1-氮雜雙環[2.2.2]辛-3-基)苯并呋喃-2-甲酰胺鹽酸鹽在玻璃小瓶中稱重并加熱到50℃。向固體中加入100μl小份的1-丁醇/水(5體積%的水)直到形成澄清溶液(總共500μl)。將樣品在50℃攪拌1小時并進行觀測。在50℃加熱1小時后,樣品仍然是澄清溶液并以1.4℃/小時的速率將其從50℃冷卻到25℃。樣品在冷卻時仍然是澄清溶液,并用帶針孔的石蠟膜覆蓋,在周圍環境溫度下允許其蒸發。在2周后,在經過部分蒸發的樣品中可見大結晶。圖13是(2S,3R)-N-(2-((3-吡啶基)甲基)-1-氮雜雙環[2.2.2]辛-3-基)苯并呋喃-2-甲酰胺一鹽酸鹽的XRPD,顯示了觀測圖樣(較亮)和計算圖樣(較暗)。實驗圖樣得自(2S,3R)-N-(2-((3-吡啶基)甲基)-1-氮雜雙環[2.2.2]辛-3-基)苯并呋喃-2-甲酰胺一鹽酸鹽的樣品,而計算實例則得自本說明書所述的單晶X射線結構,并且描述于圖10A和10B中。這兩個圖形在2θ值方面都是一致的,并且在強度和峰寬的微小差異可歸因于儀器分辨率和優選取向的影響。另外,微小差異可歸因于由于在室溫下被收集的觀察數據和收集在120K下的結構的計算數據所導致的溫度變動。甲苯磺酸鹽,特別是結晶單鹽,經過證實并且使用CuKα輻射(40kV,40mA),θ-θ測角器,V20發散度和接收狹縫,石墨二級單色器和閃爍計數器獲得的衍射圖樣如圖14所示。在40℃/75%RH下歷時1周后甲苯磺酸鹽的XRPD衍射圖揭示了改變,但是樣品仍然是1型。這一改變很可能歸因于更大程度的水合形式。VII.生物學試驗化合物A及其立體異構體結合并調節各種NNR亞型的功能的能力根據Mazurov等人的美國專利6,953,855所述進行評價,通過引用將該文獻的內容并入本說明書。化合物A的受體選擇性表征(包括5HT3受體和毒蕈堿性受體)由BiosciencesCorporation進行。在以下的兩個表達系統內進行α7NNR應答的電生理學測量:在哺乳動物GH4C1細胞中的大鼠α7NNR和在非洲爪蟾(Xenopus)卵母細胞中的人α7NNR。根據Placzek等人,Mol.Pharm.68(6):1863-1876(2005)所述,制備了表達大鼠α7NNR的GH4C1細胞,通過引用將該文獻并入本說明書。使用流體動力快速灌注系統和膜片鉗,使用該GH4C1細胞表達系統,實現了激動劑活性的電生理學測量。乙酰膽堿和煙堿都產生由α7介導的電流的濃度依賴性激活。得自文獻的激動劑EC50值與使用該方法獲得的那些EC50值是可比較的(參見Dunlop等人BiochemPharmacol付梓(2007)和Dynaflowonlinematerials(www.cellectricon.com),各自作為關于這一方法的參考被引用并入)。用Axopatch700A放大器記錄的全細胞電流在1kHz下進行過濾并使用PCI卡片(NationalInstrument)在5kHz下取樣。與先前的研究相比,根據所述來調節鹽水溶液以增加電流穩定性。在室溫下在以下的細胞外培養基中對細胞進行記錄:130mMNaCl,5mMKCl,2mMCaCl2,2mMMgCl2,10mMHEPES,用含水NaOH調節到pH7.4。將硼硅酸鹽電極(3-5MΩ)充滿以下培養基:130mMTRIS磷酸鹽,5mMNaCl,2mMMgCl2,10mMHEPES,10mMEGTA,用含水KOH調節到pH7.4(參見,Wu等人,J.Physiol.576:103-118(2006),該文獻作于關于這一教導的參考被引用并入)。在這些條件下,當用1000μM乙酰膽堿(ACh)濃度引發時,使用NNR全細胞記錄獲得的大電流活性持續長達60分鐘。采用得自Dynaflow的Cellectricon應用注釋的細胞處理程序。簡而言之,將細胞從溫育箱中取出后,將細胞用記錄介質充分洗滌三次并置于倒置Zeiss顯微鏡的平臺上。在建立全細胞記錄結構之前,平均5分鐘是必要的。為了避免改變細胞條件,將每單一細胞負載的單一細胞記錄進Dynaflow硅片中。在實驗條件下可以檢測到在應答性細胞的級分之間沒有差異。大于95%的細胞對ACh應答,并且凡是提供可測量電流的細胞都被考慮在內。在整個實驗中細胞被保持在-60mV。所有的供試制品溶液每天從儲備溶液被制得。新鮮的乙酰膽堿(ACh)儲備溶液每天在林格氏液中被制備并進行稀釋。使用Prism5.0軟件,通過單一Hill方程描繪劑量反應曲線。根據通過引用將其并入本說明書的Papke和Papke的Brit.J.Pharmacol.137:49-61(2002)所述,制備了表達人α7NNR的非洲爪蟾卵母細胞。使用成熟(>9cm)的雌性滑爪蟾(Xenopuslaevis)非洲蟾蜍(Nasco,Ft.Atkinson,WI)作為卵母細胞的來源。在手術前,通過將該動物置于1.5g/L的3-氨基苯甲酸乙酯的溶液中歷時30分鐘而將蟾蜍麻醉。從在腹部制造的切口中取出卵母細胞。為了除去卵泡細胞層,在室溫下,在不含鈣的Barth's溶液(88mMNaCl,1mMKCl,15mMHEPESpH7.6,0.81mMMgSO4,2.38mMNaHCO3,0.1mg/mL硫酸慶大霉素)中,將收獲的卵母細胞用得自WorthingtonBiochemicalCorporation(Freehold,NJ)的1.25mg/mL膠原酶處理2小時。然后,分離出5期(stage)卵母細胞并用各自為50nL(5-20ng)的人α7cRNA注射該細胞。在注射后第2-7天進行記錄。每天在林格氏液中制備新鮮的乙酰膽堿(ACh)儲備溶液。使用OpusXpress6000A(AxonInstruments,UnionCityCA)進行實驗。OpusXpress是提供最多八個并聯的卵母細胞的自動化刺穿和電壓鉗的集成系統。電壓和電流電極都充滿3M的KCl。在-60mV的保持電位下對細胞進行電壓鉗試驗。在50Hz下收集數據并在20Hz下進行過濾。將細胞用林格氏液進行浴灌注,并借助于一次性尖頭從96孔板中釋放激動劑溶液,這消除了任何交叉污染的可能性。流速被設定為2ml/min。藥物施加在ACh對照和實驗激動劑之間交替進行。施加是12秒的持續期、然后是181秒的間歇期。α7受體的應答被計算為凈電荷(參見,如上所述的Papke和Papke,Brit.J.Pharmacol.137:49-61(2002))。每個卵母細胞接受最初的ACh的對照施加,然后接受實驗藥物施加,然后接受隨后的ACh(300μM)對照施加。對實驗藥物施加的應答相對于上一次的ACh對照應答進行計算,以便對數據歸一化,補償在卵母細胞之間的變化的通道表達水平。注意,300μMACh激發得自α7受體的最大凈電荷應答,以便向ACh對照的歸一化有效地將數據歸一化到ACh最大應答。從每個實驗的濃度的至少四個卵母細胞的經歸一化的應答計算平均數和標準誤差(SEM)。對于濃度-應答關系,使用Kaleidagraph3.0.2(AbelbeckSoftware;Reading,PA)將從凈電荷分析得到的數據進行繪圖,并從Hill方程式產生曲線。根據以下規程進行化合物A的行為表征。根據通過引用將其并入本說明書的Ennaceur和Delacour的Behav.BrainRes.100:85-92(1988)的說明進行物體識別(OR)任務。根據通過引用將其并入本說明書的Levin等人的Behav.Pharm.10:675-680(1999)的說明進行放射狀臂形迷宮(RAM)模型。根據Suemaru等人的Brit.J.Pharmacol.142(5):843-850(2004)的說明進行預脈沖抑制(PPI)試驗。根據Roux等人的Curr.ProtocolsinPharmacol.Unit5.17(1999)的說明進行由阿撲嗎啡誘導的運動活力(APOLOCO)的反轉試驗。體外生物活性總結化合物A以~1nM的平衡常數(Ki)競爭性抑制放射性標記的MLA與大鼠腦海馬α7NNR的結合,顯示了其對于α7NNR亞型具有非常高的親合性。化合物A的立體異構體在大鼠α7NNR處具有以下的Ki值:2R,3S(42nM)[先前報道為28nM];2R,3R(1nM);2S,3S(25μM)(參見圖1A)。如圖1A2所示,化合物A的2S,3R對映體在α7亞型處顯示與其它三種對映體類似物相反的活性,其它三種對映體類似物作為具有弱活性的重疊點被提供。化合物A不具有任何顯著的親合性(Ki值>2μM)地結合于α4β2NNR。使用在GH4C1(哺乳動物)細胞中穩定表達的大鼠α7NNR進行的膜片鉗電生理學技術考察了(2S,3R)-N-(2-((3-吡啶基)甲基)-1-氮雜雙環[2.2.2]辛-3-基)苯并呋喃-2-甲酰胺或其藥學上可接受的鹽(化合物A)及其立體異構體的功能活性。在這些實驗中,化合物A與其它單獨的異構體和所有四種異構體的外消旋混合物相比,產生了顯著不同的功能性質。從圖1A和1B可見,化合物A在引發功能應答方面(Emax=93%,相對于乙酰膽堿(ACh);EC50=14nM)比任一種其它的異構體或四種異構體的混合物具有更強的功效和更有效。實際上,化合物A(2S,3R異構體)是N-(2-((3-吡啶基)甲基)-1-氮雜雙環[2.2.2]辛-3-基)苯并呋喃-2-甲酰胺的唯一的能夠在1-50nM的整個濃度范圍內提供強效的激動作用的異構體,采用與本說明書所述的體內活性有關的10nM,如圖1B所示。化合物A的功能活性還在瞬時表達人α7NNR的非洲爪蟾卵母細胞中進行了電生理學評價。在該系統中,化合物A具有33nM的EC50值和100%ACh應答的Emax。在施用濃度大于100nM(IC50=200nM)的(2S,3R)-N-(2-((3-吡啶基)甲基)-1-氮雜雙環[2.2.2]辛-3-基)苯并呋喃-2-甲酰胺后的隨后的對ACh的對照應答中有增加。與先前所述的α7完全激動劑(參見Astles等人,CurrentDrugTargetsCNSNeurologicalDisorders1(4):337-348(2002),其作為關于這一報道的參考被并入本說明書)相反,(2S,3R)-N-(2-((3-吡啶基)甲基)-1-氮雜雙環[2.2.2]辛-3-基)苯并呋喃-2-甲酰胺的EC50和IC50之間的差別表明產生α7的半數最大功能應答的濃度導致最小的而非完全的殘余抑制。當將(2S,3R)-N-(2-((3-吡啶基)甲基)-1-氮雜雙環[2.2.2]辛-3-基)苯并呋喃-2-甲酰胺施用于表達人α4β2亞型的卵母細胞時沒有可檢測的激活,并且在隨后的對ACh的對照應答中沒有顯著降低,表明(2S,3R)-N-(2-((3-吡啶基)甲基)-1-氮雜雙環[2.2.2]辛-3-基)苯并呋喃-2-甲酰胺在α4β2處既不是激動劑也不是拮抗劑。化合物在攜帶肌肉型受體(在人TE671/RD克隆細胞中的α1β1γδ亞型)、或神經節型受體(在大鼠嗜鉻細胞瘤PC12細胞中和在人SHSY-5Y克隆細胞中的α3β4亞型)的功能模型中表現出很少的激動劑活性或不表現出激動劑活性,在這些亞型處產生≤10%(人肌肉),≤20%(大鼠神經節)和≤10%(人神經節)的煙堿應答。這些數據顯示了對CNS亞型的選擇性高于對PNS亞型的選擇性。由于在α7和5-羥色胺(5HT3)受體之間的序列接近和結構同源性,以及使用其它的煙堿樣配體觀察到的對這兩種受體的交叉反應性,考察了化合物A對5HT3受體的親合性。化合物A(10μM)顯示了在小鼠5HT3受體處的放射性配體結合的59%抑制和在人受體處的放射性配體結合的25%抑制。對在人5HT3受體處的功能激活的考察暗示了最小激活到無激活(即,在100μM得到15%激活的最大應答)。毒蕈堿性受體由于已經觀察到的與其它煙堿樣配體的相互作用而成為另一個有意義的領域。當在對M1、M2、非選擇性中心毒蕈堿性受體和非選擇性外周毒蕈堿性受體的競爭性結合抑制試驗中進行考察時,化合物A顯示最小的相互作用到無相互作用。數據顯示化合物A對α7NNR配體具有選擇性。化合物A在周圍神經系統所特有的煙堿樣受體的這些亞型處或在毒蕈堿性受體或5HT35-羥色胺能受體處不發生良好結合。因此,化合物A在治療中樞神經系統障礙中具有治療潛力,并且不產生與周圍神經系統的相互作用有關的副作用。體內生物活性總結化合物A(2S,3R)-N-(2-((3-吡啶基)甲基)-1-氮雜雙環[2.2.2]辛-3-基)苯并呋喃-2-甲酰胺或其藥學上可接受的鹽在兩個認知行為模型中顯示顯著的功效。化合物A在大鼠的物體識別模型中,在i.p.(腹膜內給藥,圖3)和p.o.(口服給藥,圖4)后顯示強效的活性,還在口服給藥后的寬的劑量范圍內顯示活性(圖4)。在相同的低劑量(0.3和1mg/kg)下腹膜內給藥的化合物A傾向于反轉在OR任務中由MK-801誘導的缺陷(圖5),并且以0.3mg/kg被口服給藥的化合物A具有持續至少18小時的認知作用(圖6)。在考查工作記憶的放射狀臂形迷宮(RAM)(圖7)模型中,化合物A顯著增加在錯誤選擇之前的正確選擇數。這些結果顯示了(2S,3R)-N-(2-((3-吡啶基)甲基)-1-氮雜雙環[2.2.2]辛-3-基)苯并呋喃-2-甲酰胺在治療與精神分裂癥有關的認知缺陷和功能障礙(包括與工作記憶有關的功能障礙)方面具有潛力。對于適用于治療精神分裂癥中的認知功能障礙的化合物,其不可削弱對抗精神分裂癥的陽性癥狀的典型性或非典型性抗精神病藥的影響。因此,除了化合物A的認知增強性質之外,令人注目的是,化合物A還有效地反轉由阿撲嗎啡誘導的運動活力(APOLOCO)(圖8),并在精神分裂癥的陽性癥狀的預脈沖抑制(PPI)(圖9)模型中有效。因此,(2S,3R)-N-(2-((3-吡啶基)甲基)-1-氮雜雙環[2.2.2]辛-3-基)苯并呋喃-2-甲酰胺將被預期在對抗與精神分裂癥有關的陽性癥狀以及認知癥狀中提供額外的益處。觀察到的特定的藥理學應答可根據或取決于被選擇的具體活性化合物,或者是否存在藥學載體,以及所采用的制劑類型和給藥方式而變化,并且根據本發明的實踐,在結果中的這些被預期的變化或差異被本發明所涵蓋。盡管已經說明和詳細描述了本發明的具體的實施方案,但是本發明不限于這些具體的實施方案。上述的詳細描說明作為本發明的示例而被提供,不應認為對本發明構成任何的限制。修改對本領域技術人員來說是明顯的,并且這些修飾都不脫離本發明的精神,并且意在被所附權利要求書的范圍所包括。當前第1頁1 2 3
相關技術
網友詢問留言
已有0條留言
- 還沒有人留言評論。精彩留言會獲得點贊!
1