一種新型氟硼酸類化合物的放射性標(biāo)記方法與流程
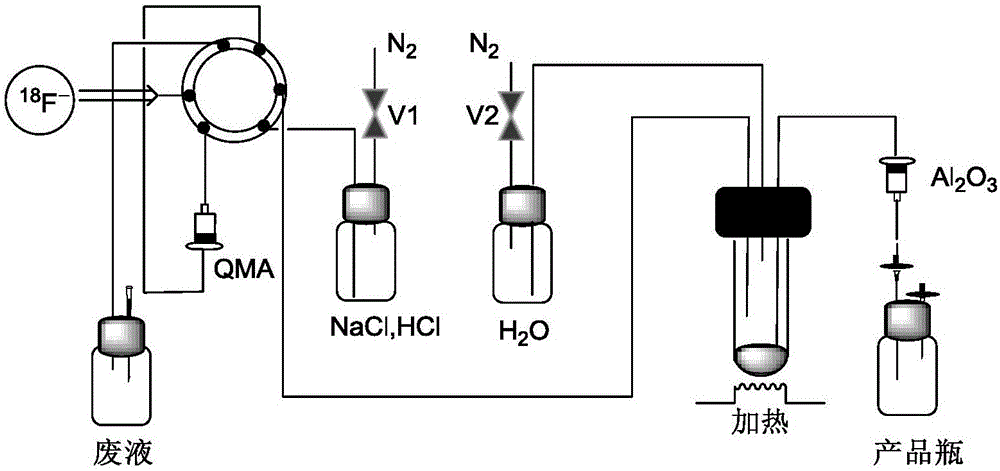
本發(fā)明屬于醫(yī)學(xué)影像領(lǐng)域,涉及氟硼酸類化合物的放射性標(biāo)記方法,具體涉及一種氟硼酸類化合物的[18F]放射性標(biāo)記方法。
背景技術(shù):
氨基酸是腫瘤增殖過程的必需營養(yǎng)物質(zhì),對氨基酸類化合物進(jìn)行放射性標(biāo)記以實(shí)現(xiàn)腫瘤的特異性顯像是近些年的研究熱點(diǎn)。只是由于氨基酸結(jié)構(gòu)的特殊性,傳統(tǒng)的放射性標(biāo)記方法如Al18F、68Ga、99mTc等都會(huì)對原有分子結(jié)構(gòu)進(jìn)行比較大的改變,容易導(dǎo)致標(biāo)記分子的體內(nèi)代謝行為出現(xiàn)差異。
氟硼酸結(jié)構(gòu)是一種與α-氨基酸結(jié)構(gòu)極其類似的結(jié)構(gòu),研究表明用氟硼酸結(jié)構(gòu)取代α-氨基酸結(jié)構(gòu)后化合物的藥代行為與原來氨基酸非常相似,因此可以用氟硼酸類化合物代替氨基酸類化合物進(jìn)行放射性標(biāo)記用于腫瘤的顯像研究。
傳統(tǒng)的氟硼酸類化合物放射性標(biāo)記方法采用氨基聚醚碳酸鉀溶液進(jìn)行淋洗,安全系數(shù)不高,且最后需要檢查氨基聚醚和乙腈等有害物質(zhì)的殘留,工藝復(fù)雜;傳統(tǒng)的C18柱固相萃取柱純化氟硼酸類化合物也存在耗時(shí)長、產(chǎn)品損失大,操作工序復(fù)雜等問題。
技術(shù)實(shí)現(xiàn)要素:
為克服上述現(xiàn)有技術(shù)的缺陷,本發(fā)明的目的在于提供一種新型氟硼酸類化合物的放射性標(biāo)記方法,采用放射性[18F]對氟硼酸類化合物進(jìn)行放射性標(biāo)記、分離和純化。
本發(fā)明的上述目的通過以下技術(shù)方案實(shí)現(xiàn):
一種新型氟硼酸類化合物的放射性標(biāo)記方法,包括以下步驟:
(1)采用質(zhì)子回旋加速器進(jìn)行18O(p,n)18F反應(yīng)生成18F-;
(2)將步驟(1)中所述18F-經(jīng)管道傳輸至陰離子交換柱QMA上,同時(shí)被所述QMA捕獲;
(3)開啟氮?dú)忾yV1,步驟(2)中所述QMA經(jīng)N2吹干后,用pH=2、濃度為0.01mol/L的鹽酸氯化鈉溶液0.5mL將所述18F-淋洗至反應(yīng)瓶中;
(4)在90℃的加熱條件下,將濃度為500mmol/L的[19F]氟硼酸類化合物的乙醇溶液0.02mL與步驟(3)中所述淋洗后的18F-進(jìn)行離子反應(yīng);20min后開啟氮?dú)忾yV2,在N2保護(hù)下向上述離子交換反應(yīng)體系中加入去離子水5mL進(jìn)行淬滅反應(yīng);
(5)將步驟(4)中淬滅反應(yīng)后的體系直接過中性Al2O3柱純化后滴入裝有pH=7的5mL磷酸氫二鈉-檸檬酸緩沖液的反應(yīng)瓶中,濾液即為[18F]氟硼酸類化合物溶液,然后將所述[18F]氟硼酸類化合物溶液進(jìn)行質(zhì)控分析。
步驟(1)中所述的質(zhì)子回旋加速器包括GE公司Qilin醫(yī)用質(zhì)子回旋加速器,所述加速器束流為30μA,轟擊時(shí)間為10min。
步驟(2)中所述的18F-放射性活度為150-200mCi。
步驟(3)中所述的QMA殘留18F-放射性活度為2.0-3.0mCi。
步驟(4)中所述的[19F]氟硼酸類化合物與所述的18F-通過離子交換與硼原子共價(jià)鍵連接。
步驟(4)中所述的[19F]氟硼酸類化合物包含硼原子上連有一個(gè)、兩個(gè)或者三個(gè)氟原子的化合物。
步驟(4)中所述的[19F]氟硼酸類化合物如下式(1)所示;步驟(5)中所述的[18F]氟硼酸類化合物如下式(2)所示;步驟(4)中所述的氟硼酸類化合物的核素交換反應(yīng)如下式(3)所示。
步驟(5)中所述的[18F]氟硼酸類化合物放射性活度為10-20mCi。
步驟(5)中所述的質(zhì)控分析包括色澤、澄清度、pH、放化純度、細(xì)菌內(nèi)毒素。
本發(fā)明具有如下優(yōu)點(diǎn):
1.本發(fā)明首次提出用鹽酸氯化鈉溶液洗脫18F-,既避免了為保證反應(yīng)濃度對反應(yīng)18F-的濃縮,也避免了將18F-傳輸至濃縮管再轉(zhuǎn)至反應(yīng)管過程中的損失;同時(shí),采用鹽酸氯化鈉溶液作為淋洗18F-溶液,不僅可以極好的保證對18F-的淋洗效率,還保證反應(yīng)過程中的酸性要求。
2.相比傳統(tǒng)的氨基聚醚碳酸鉀溶液,本申請的淋洗18F-溶液鹽酸氯化鈉溶液對人體幾乎無毒副作用,無需額外純化步驟,經(jīng)簡單的稀釋即可直接用于人體,安全系數(shù)高,而且產(chǎn)品無需檢查氨基聚醚和乙腈等有機(jī)物的殘留。
3.傳統(tǒng)的C18柱固相萃取純化氟硼酸類化合物工藝耗時(shí)長、產(chǎn)品回收率低,采用本發(fā)明標(biāo)記反應(yīng)的純化方法只需簡單加緩沖液稀釋后過Al2O3柱吸附游離的18F-,濾液即可作為注射液使用,操作簡單,純化快速,產(chǎn)品收率高。
附圖說明
圖1為本申請的一種新型氟硼酸類化合物的放射性標(biāo)記工藝流程圖。
具體實(shí)施方式
以下結(jié)合附圖通過實(shí)施例1-5和對比例1-3來說明本發(fā)明,但不用來限制本發(fā)明的范圍。
實(shí)施例1
一種新型氟硼酸類化合物的放射性標(biāo)記方法,包括以下步驟:
(1)采用GE公司Qilin醫(yī)用質(zhì)子回旋加速器進(jìn)行18O(p,n)18F反應(yīng)生成18F-,加速器束流為30μA,轟擊時(shí)間為10min;
(2)將步驟(1)中18F-經(jīng)管道傳輸至Sep-Park Light QMA上,18F-被QMA捕獲,18F-放射性活度為155mCi;
(3)開啟氮?dú)忾yV1,步驟(2)中QMA經(jīng)N2吹干后,用pH=2、濃度為0.01mol/L的鹽酸氯化鈉溶液0.5mL將所述18F-淋洗至反應(yīng)瓶中,QMA殘留的18F-放射性活度為2.2mCi;
(4)在90℃的加熱條件下,將濃度為500mmol/L的[19F]苯丙氟硼酸乙醇溶液0.02mL與步驟(3)中淋洗后的18F-進(jìn)行離子反應(yīng);20min后開啟氮?dú)忾yV2,向上述離子交換反應(yīng)體系中加入去離子水5mL進(jìn)行淬滅反應(yīng);
(5)將步驟(4)中淬滅反應(yīng)后的體系直接過Sep-Park Light Al2O3柱純化后滴入裝有pH=7的5mL磷酸氫二鈉-檸檬酸緩沖液的反應(yīng)瓶中,濾液即為[18F]苯丙氟硼酸溶液,放射性活度為16.2mCi;然后將該產(chǎn)品溶液進(jìn)行質(zhì)控分析。
產(chǎn)品質(zhì)控分析結(jié)果表明,產(chǎn)品溶液澄清透明,pH為6.3,HPLC測定的放化純度為99%,無細(xì)菌內(nèi)毒素,可直接作為注射液使用。
實(shí)施例2
(1)采用GE公司Qilin醫(yī)用質(zhì)子回旋加速器進(jìn)行18O(p,n)18F反應(yīng)生成18F-,加速器束流為30μA,轟擊時(shí)間為10min;
(2)將步驟(1)中18F-經(jīng)管道傳輸至Sep-Park Light QMA上,18F-被QMA捕獲,18F-放射性活度為163mCi;
(3)開啟氮?dú)忾yV1,步驟(2)中QMA經(jīng)N2吹干后,用pH=2、濃度為0.01mol/L的鹽酸氯化鈉溶液0.5mL將所述18F-淋洗至反應(yīng)瓶中,QMA殘留的18F-放射性活度為2.5mCi;
(4)在90℃的加熱條件下,將濃度為500mmol/L的[19F]苯丙氟硼酸乙醇溶液0.02mL與步驟(3)中淋洗后的18F-進(jìn)行離子反應(yīng);20min后開啟氮?dú)忾yV2,向上述離子交換反應(yīng)體系中加入去離子水5mL進(jìn)行淬滅反應(yīng);
(5)將步驟(4)中淬滅反應(yīng)后的體系直接過Sep-Park Light Al2O3柱純化后滴入裝有pH=7的5mL磷酸氫二鈉-檸檬酸緩沖液的反應(yīng)瓶中,濾液即為[18F]苯丙氟硼酸溶液,放射性活度為15.8mCi;然后將該產(chǎn)品溶液進(jìn)行質(zhì)控分析。
產(chǎn)品質(zhì)控分析結(jié)果表明,產(chǎn)品溶液澄清透明,pH為6.5,HPLC測定的放化純度為99%,無細(xì)菌內(nèi)毒素,可直接作為注射液使用。
實(shí)施例3
一種新型氟硼酸類化合物的放射性標(biāo)記方法,包括以下步驟:
(1)采用GE公司Qilin醫(yī)用質(zhì)子回旋加速器進(jìn)行18O(p,n)18F反應(yīng)生成18F-,加速器束流為30μA,轟擊時(shí)間為10min;
(2)將步驟(1)中18F-經(jīng)管道傳輸至Sep-Park Light QMA上,18F-被QMA捕獲,18F-放射性活度為171mCi;
(3)開啟氮?dú)忾yV1,步驟(2)中QMA經(jīng)N2吹干后,用pH=2、濃度為0.01mol/L的鹽酸氯化鈉溶液0.5mL將所述18F-淋洗至反應(yīng)瓶中,QMA殘留的18F-放射性活度為2.7mCi;
(4)在90℃的加熱條件下,將濃度為500mmol/L的[19F]苯丙氟硼酸乙醇溶液0.02mL與步驟(3)中淋洗后的18F-進(jìn)行離子反應(yīng);20min后開啟氮?dú)忾yV2,向上述離子交換反應(yīng)體系中加入去離子水5mL進(jìn)行淬滅反應(yīng);
(5)將步驟(4)中淬滅反應(yīng)后的體系直接過Sep-Park Light Al2O3柱純化后滴入裝有pH=7的5mL磷酸氫二鈉-檸檬酸緩沖液的反應(yīng)瓶中,濾液即為[18F]苯丙氟硼酸溶液,放射性活度為16.5mCi;然后將該產(chǎn)品溶液進(jìn)行質(zhì)控分析。
產(chǎn)品質(zhì)控分析結(jié)果表明,產(chǎn)品溶液澄清透明,pH為6.6,HPLC測定的放化純度為99%,無細(xì)菌內(nèi)毒素,可直接作為注射液使用。
實(shí)施例4
一種新型氟硼酸類化合物的放射性標(biāo)記方法,包括以下步驟:
(1)采用GE公司Qilin醫(yī)用質(zhì)子回旋加速器進(jìn)行18O(p,n)18F反應(yīng)生成18F-,加速器束流為30μA,轟擊時(shí)間為10min;
(2)將步驟(1)中18F-經(jīng)管道傳輸至陰離子交換柱QMA上,同時(shí)被所述QMA捕獲,18F-放射性活度為200mCi;
(3)開啟氮?dú)忾yV1,步驟(2)中QMA經(jīng)N2吹干后,用pH=2、濃度為0.01mol/L的鹽酸氯化鈉溶液0.5mL將所述18F-淋洗至反應(yīng)瓶中,QMA殘留18F-放射性活度為2.5mCi;
(4)在90℃的加熱條件下,將濃度為500mmol/L的[19F]苯丙氟硼酸乙醇溶液0.02mL與步驟(3)中淋洗后的18F-進(jìn)行離子反應(yīng);20min后開啟氮?dú)忾yV2,在N2保護(hù)下向上述離子交換反應(yīng)體系中加入去離子水5mL淬滅反應(yīng);
(5)將步驟(4)中淬滅反應(yīng)后的體系直接過中性Al2O3柱純化后滴入裝有pH=7的5mL磷酸氫二鈉-檸檬酸緩沖液的反應(yīng)瓶中,濾液即為[18F]苯丙氟硼酸溶液,放射性活度為18mCi;然后將該產(chǎn)品溶液進(jìn)行質(zhì)控分析。
產(chǎn)品質(zhì)控分析結(jié)果表明,產(chǎn)品溶液澄清透明,pH為6.4,HPLC測定的放化純度為99%,無細(xì)菌內(nèi)毒素,可直接作為注射液使用。
實(shí)施例5
一種新型氟硼酸類化合物的放射性標(biāo)記方法,包括以下步驟:
(1)采用GE公司Qilin醫(yī)用質(zhì)子回旋加速器進(jìn)行18O(p,n)18F反應(yīng)生成18F-,加速器束流為30μA,轟擊時(shí)間為10min;
(2)將步驟(1)中18F-經(jīng)管道傳輸至陰離子交換柱QMA上,同時(shí)被所述QMA捕獲,18F-放射性活度為150mCi;
(3)開啟氮?dú)忾yV1,步驟(2)中QMA經(jīng)N2吹干后,用pH=2、濃度為0.01mol/L的鹽酸氯化鈉溶液0.5mL將所述18F-淋洗至反應(yīng)瓶中,QMA殘留18F-放射性活度為2.0mCi;
(4)在90℃的加熱條件下,將濃度為500mmol/L的[19F]氟硼酸類化合物的乙醇溶液0.02mL與步驟(3)中淋洗后的18F-進(jìn)行離子反應(yīng);20min后開啟氮?dú)忾yV2,在N2保護(hù)下向上述離子交換反應(yīng)體系中加入去離子水5mL淬滅反應(yīng);
(5)將步驟(4)中淬滅反應(yīng)后的體系直接過中性Al2O3柱純化后滴入裝有pH=7的5mL磷酸氫二鈉-檸檬酸緩沖液的反應(yīng)瓶中,濾液即為[18F]氟硼酸類化合物溶液,放射性活度為11mCi;然后將該產(chǎn)品溶液進(jìn)行質(zhì)控分析。
產(chǎn)品質(zhì)控分析結(jié)果表明,產(chǎn)品溶液澄清透明,pH為6.5,HPLC測定的放化純度為99%,無細(xì)菌內(nèi)毒素,可直接作為注射液使用。
對比例1
采用對比文獻(xiàn)和專利文獻(xiàn)中記載氟硼酸類化合物的放射性標(biāo)記方法,包括以下步驟:
(1)采用GE公司Qilin醫(yī)用質(zhì)子回旋加速器進(jìn)行18O(p,n)18F反應(yīng)生成18F-,加速器束流為30μA,轟擊時(shí)間為10min;
(2)將步驟(1)中18F-經(jīng)管道傳輸至Sep-Park Light QMA上,同時(shí)被QMA捕獲,18F-放射性活度為171mCi;
(3)反應(yīng)管中加入0.02mL濃度為500mmol/L的苯丙氟硼酸乙醇溶液和0.2mL pH為2.5的噠嗪-鹽酸緩沖液;用K2.2.2、K2CO3的混合溶液0.5mL將步驟(2)中的18F-淋洗至反應(yīng)瓶中,QMA殘留的18F-放射性活度為2.4mCi;
(4)在60℃的加熱條件下,[19F]苯丙氟硼酸溶液與步驟(3)中淋洗后的18F-進(jìn)行核素交換反應(yīng);20min后向上述核素交換反應(yīng)體系加入去離子水5mL進(jìn)行淬滅反應(yīng);
(5)將步驟(4)中淬滅反應(yīng)后的體系直接過Sep-Park Light C18柱純化,再用10mL水清洗C18柱,C18柱上捕獲產(chǎn)品11.3mCi,然后用50%的乙醇溶液5mL淋洗C18柱即得放射性活度為7.2mCi的[18F]苯丙氟硼酸溶液,C18柱殘留18F-放射性活度為3.6mCi,最后對該[18F]苯丙氟硼酸溶液進(jìn)行質(zhì)控分析。
產(chǎn)品質(zhì)控分析結(jié)果表明,產(chǎn)品溶液澄清透明,pH為7,HPLC測定的放化純度為99%,無細(xì)菌內(nèi)毒素,乙醇含量為0.0149%,乙腈含量為0.01%,K2.2.2含量達(dá)標(biāo)。
對比例2
采用對比文獻(xiàn)和專利文獻(xiàn)中記載氟硼酸類化合物的放射性標(biāo)記方法,包括以下步驟:
(1)采用GE公司Qilin醫(yī)用質(zhì)子回旋加速器進(jìn)行18O(p,n)18F反應(yīng)生成18F-,加速器束流為30μA,轟擊時(shí)間為10min;
(2)將步驟(1)中18F-經(jīng)管道傳輸至Sep-Park Light QMA上,同時(shí)被QMA捕獲,18F-放射性活度為175mCi;
(3)反應(yīng)管中加入0.02mL濃度為500mmol/L的苯丙氟硼酸乙醇溶液和0.2mL pH為2.5的噠嗪-鹽酸緩沖液;用K2.2.2、K2CO3的混合溶液0.5mL將步驟(2)中的18F-淋洗至反應(yīng)瓶中,QMA殘留的18F-放射性活度為2.1mCi;
(4)在60℃的加熱條件下,[19F]苯丙氟硼酸溶液與步驟(3)中淋洗后的18F-進(jìn)行核素交換反應(yīng);20min后向上述核素交換反應(yīng)體系加入去離子水5mL進(jìn)行淬滅反應(yīng);
(5)將步驟(4)中淬滅反應(yīng)后的體系直接過Sep-Park Light C18柱純化,再用10mL水清洗C18柱,C18柱上捕獲產(chǎn)品12.1mCi,然后用50%的乙醇溶液5mL淋洗C18柱即得放射性活度為7.5mCi的[18F]苯丙氟硼酸溶液,C18柱殘留18F-放射性活度為3.3mCi,最后對該[18F]苯丙氟硼酸溶液進(jìn)行質(zhì)控分析。
產(chǎn)品質(zhì)控分析結(jié)果表明,產(chǎn)品溶液澄清透明,pH為7,HPLC測定的放化純度為99%,無細(xì)菌內(nèi)毒素,乙醇含量為0.006%,乙腈含量為0.012%,K2.2.2含量達(dá)標(biāo)。
對比例3
采用對比文獻(xiàn)和專利文獻(xiàn)中記載氟硼酸類化合物的放射性標(biāo)記方法,包括以下步驟:
(1)采用GE公司Qilin醫(yī)用質(zhì)子回旋加速器進(jìn)行18O(p,n)18F反應(yīng)生成18F-,加速器束流為30μA,轟擊時(shí)間為10min;
(2)將步驟(1)中18F-經(jīng)管道傳輸至Sep-Park Light QMA上,同時(shí)被QMA捕獲,18F-放射性活度為168mCi;
(3)反應(yīng)管中加入0.02mL濃度為500mmol/L的苯丙氟硼酸乙醇溶液和0.2mL pH為2.5的噠嗪-鹽酸緩沖液;用K2.2.2、K2CO3的混合溶液0.5mL將步驟(2)中的18F-淋洗至反應(yīng)瓶中,QMA殘留的18F-放射性活度為2.0mCi;
(4)在60℃的加熱條件下,[19F]苯丙氟硼酸溶液與步驟(3)中淋洗后的18F-進(jìn)行核素交換反應(yīng);20min后向上述核素交換反應(yīng)體系加入去離子水5mL進(jìn)行淬滅反應(yīng);
(5)將步驟(4)中淬滅反應(yīng)后的體系直接過Sep-Park Light C18柱純化,再用10mL水清洗C18柱,C18柱上捕獲產(chǎn)品10.8mCi,然后用50%的乙醇溶液5mL淋洗C18柱即得放射性活度為6.5mCi的[18F]苯丙氟硼酸溶液,C18柱殘留18F-放射性活度為3.8mCi,最后對該[18F]苯丙氟硼酸溶液進(jìn)行質(zhì)控分析。
產(chǎn)品質(zhì)控分析結(jié)果表明,產(chǎn)品溶液澄清透明,pH為7,HPLC測定的放化純度為99%,無細(xì)菌內(nèi)毒素,乙醇含量為0.005%,乙腈含量為0.004%,K2.2.2含量達(dá)標(biāo)。
通過上述實(shí)施例1-5與對比例1-3可知:
1、本發(fā)明采用鹽酸氯化鈉溶液洗脫18F-,既避免為保證反應(yīng)濃度對反應(yīng)18F-的濃縮,也避免將18F-傳輸至濃縮管再轉(zhuǎn)至反應(yīng)管過程中的損失;采用鹽酸氯化鈉溶液作為淋洗18F-溶液,不僅可以極好的保證對18F-的淋洗效率,還保證反應(yīng)過程中的酸性要求。
2.與對比例1-3的放射性標(biāo)記方法相比,本申請的淋洗18F-溶液鹽酸氯化鈉溶液對人體幾乎無毒副作用,無需額外純化步驟,經(jīng)簡單的稀釋即可直接用于人體,安全系數(shù)高,而且產(chǎn)品無需檢查氨基聚醚和乙腈等有機(jī)物的殘留。
3.對比例1-3采用傳統(tǒng)的C18柱固相萃取純化氟硼酸類化合物工藝耗時(shí)長、產(chǎn)品回收率低,采用本發(fā)明標(biāo)記反應(yīng)的純化方法只需簡單加pH=7的5mL磷酸氫二鈉-檸檬酸緩沖液稀釋后過Al2O3柱吸附游離的18F-,濾液即可作為注射液使用,操作簡單,純化快速,產(chǎn)品收率高。
雖然,上文中已經(jīng)用一般性說明及具體實(shí)施例對本發(fā)明作了詳盡的描述,但在本發(fā)明基礎(chǔ)上,可以對之作一些修改或改進(jìn),這對本領(lǐng)域技術(shù)人員而言是顯而易見的。因此,在不偏離本發(fā)明精神的基礎(chǔ)上所做的這些修改或改進(jìn),均屬于本發(fā)明要求保護(hù)的范圍。
- 還沒有人留言評論。精彩留言會(huì)獲得點(diǎn)贊!
- 光可斷裂的熒光標(biāo)記化合物及用途的制作方法與工藝
- 一種氟?18標(biāo)記的化合物及其制備方法與應(yīng)用與流程
- 一種核素標(biāo)記的靶向探針化合物及其制備方法與流程
- 抗癌劑感受性的判定標(biāo)記的制造方法與工藝
- 經(jīng)放射性鹵素標(biāo)記的吡啶并[1,2?a]苯并咪唑衍生物化合物的制造方法與工藝
- 一種放射性核素碘標(biāo)記的熒光碳點(diǎn)、合成方法和應(yīng)用與流程
- 用于生物制品的18F?放射性標(biāo)記的方法和組合物與流程
- 反應(yīng)性標(biāo)記化合物及其用途的制造方法與工藝
- 一種自旋標(biāo)記駱駝寧堿A化合物、制備方法及其用途與制造工藝
- 一種新型氟硼酸類化合物的放射性標(biāo)記方法與制造工藝
- 一種EphB4 受體靶向多肽及其應(yīng)用
- 使用放射性核素標(biāo)記的膜聯(lián)蛋白診斷和治療阿爾茨海默病的制作方法
- 一種锝-99m標(biāo)記的秋水仙素配合物及其制備方法與用圖
- 基于聚乙二醇化聚乙烯亞胺的不同表面修飾的spect/ct雙模態(tài)成像造影劑的制備方法
- 多層錯(cuò)位耦合準(zhǔn)直器、輻射器、探測裝置及掃描設(shè)備的制造方法
- 放射性標(biāo)記方法
- 放射性標(biāo)記材料的制作方法
- 放射源的位置調(diào)節(jié)方法及系統(tǒng)的制作方法
- Pet多床位圖像的重建方法及裝置、合并方法及裝置的制造方法
- 基于CendR內(nèi)化增效序列的放射性核素標(biāo)記腫瘤靶向分子影像探針及其合成方法與應(yīng)用