一種高純度曲克蘆丁及其制備方法與流程
本發明涉及藥物化學和有機合成化學技術領域,具體而言,涉及一種高純度曲克蘆丁及其制備方法。
背景技術:
曲克蘆丁(Troxerutin)又稱維腦路通,三羥乙基蘆丁。化學名為7,3’,4’-三羥乙基蘆丁(簡稱三羥乙基蘆丁)。分子式C33H42O19;分子量;742.69;CAS NO:7085-55-4;化學結構式如下:
曲克蘆丁是一種抗凝血及溶栓藥,臨床通常以口服或注射給藥。國家新版藥典(2015年版)規定曲克蘆丁原料藥含7,3’,4’-三羥乙基蘆丁(C33H42O19)不得低于80%(口服級)和88.0%(注射級)。作為注射級原料藥,對其半合成過程中的同分異構體一、二和四羥乙基蘆丁的峰面積之和、單個未知雜質峰面積、未知雜質總峰面積以及與總峰面積的百分比均有明確而嚴格的限定。
作為注射級原料藥,毋庸置疑,其活性成分三羥乙基蘆丁含量越高,療效越高,且副作用越小。
曲克蘆丁原料藥的制備條件盡管有差異,但均是以蘆丁和環氧乙烷為原料,以水和/或甲醇、乙醇為反應介質,在堿性催化劑存在下發生Willanson醚化親核取代反應(簡稱醚化反應或羥乙基化反應),生成以7,3’,4’-三羥乙基蘆丁為主,同時伴有一定量的一、二和四羥乙基蘆丁異構體及少量未知雜質組分的生成。事實上,至反應后期,三羥乙基和四羥乙基蘆丁近乎同步增長。換言之,反應越徹底,四羥乙基蘆丁的生成比例越高。反之,則有少量二羥乙基和一羥乙基蘆丁異構體不能繼續轉化為目標產物。
雖然四羥乙基蘆丁的水溶性最好,但其活性最低,而且在后續分離、純化過程中難以除盡,同時在后續純化過程中還會伴隨三羥乙基蘆丁產品的流失。一羥乙基和二羥乙基蘆丁雖有活性,但水溶性不及三羥乙基蘆丁,從而直接影響曲克蘆丁注射液的澄清度,并由此產生可能的臨床不良反應。
在已見報道的各種醚化反應及后續分離、純化工藝條件下,終產品中7,3’,4’-三羥乙基蘆丁的色譜純度很難超過93.0%。
曲克蘆丁的醚化反應依據反應溶劑、催化劑和反應條件的不同而形成了大同小異的多種工藝條件。如(1)以水、甲醇或甲醇-水混合溶劑作為反應溶劑的文獻或專利為主流工藝:(1)加壓反應條件:CN102924546A;(2)以不同催化劑完成的醚化反應,如:專利CN1331697A(US6855697B1)、專利CN1554353A;(3)用樹脂代替鹽酸控制反應液的PH值的控制方法,如CN1814613A;(4)采用相轉移催化醚化反應方法,CN103601774A。而利用上述方法所得到的目標產品——7,3’,4’-三羥乙基蘆丁的純度均未超過91%。顯而易見,目標產品中三羥乙基蘆丁純度越高,制備技術難度越大,產率損失也大。雖然使用色譜分離或大孔樹脂吸附分離(如:CN 104177461A)的方法可以獲得純度為98%乃至99.0%的三羥乙基蘆丁,但由于方法本身特性,所以只能用于制備標準品,而難以應用到工業生產上。
為了限制醚化反應過程中四羥乙基蘆丁異構物的生成量,河北聯合大學(CN103113437A)公開了一種曲克蘆丁的制備方法,但從其實施例1~實施例3的終產品分析結果(三羥乙基蘆丁純度:91~92%;四羥乙基蘆丁:6~7%;二羥乙基蘆丁:2~3%)來看,該方法對四羥乙基蘆丁的抑制效果仍然較差。出于相同目的,李思、梁旭彤等發明了一種有別于上述專利方法的新方法(三羥乙基蘆丁的制備方法,CN104177461A),但該中間轉化的方法和后續操作(如離子交換樹脂除鹽、大孔樹脂吸附等)工藝過于繁瑣,周期長、成本高,缺乏經濟實用性,故通過該方法制備98.0%以上的三羥乙基蘆丁的原料成本高達每千克2000~5000元,而目前國內藥用曲克蘆丁的市場價卻低于甚至遠低于每千克2000元,從生產角度考慮,其顯然缺乏生產價值,故而難以進行工業化生產。
技術實現要素:
本發明的目的在于提供一種高純度曲克蘆丁的制備方法,此制備方法適用于工業生產,成本也較低,能夠制得純度非常高的曲克蘆丁。
本發明的另一目的在于提供一種高純度曲克蘆丁。
本發明解決其技術問題是采用以下技術方案來實現的:
一種高純度曲克蘆丁的制備方法,包括:
前段醚化步驟:將蘆丁與環氧乙烷在醇、水、堿性催化劑的條件下進行醚化反應,并保持反應液中四羥乙基蘆丁的峰面積百分比在1.5%以下;其中,蘆丁、前段醚化步驟中的環氧乙烷、醇、水、堿性催化劑的質量比為1:0.1~0.4:1.5~6.5:0.5~8.0:0.008~0.012,醚化反應的反應溫度為60~80℃;
絡合步驟:調節反應溫度為70~90℃,然后在保護氣體的保護下往反應液中加入飽和的醋酸鹽的水溶液,并在加入期間控制反應液的pH為8.0~9.5,加畢,攪拌10~20min;其中,保護氣體選自氮氣、惰性氣體中的一種或多種,醋酸鹽為金屬離子醋酸鹽,蘆丁與醋酸鹽的摩爾比為1:1.0~1.3;
后段醚化步驟:調節反應溫度為60~80℃、反應液的pH為8.0~9.0,繼續通入環氧乙烷,反應至反應液中三羥乙基蘆丁的峰面積百分比不再增長時,停止反應,得到反應產物;其中,蘆丁與后續醚化步驟中的環氧乙烷的質量比為1:0.1~0.3;
后處理步驟:利用稀酸與反應產物發生水解反應,然后濃縮、精制。
另外,一種高純度曲克蘆丁,是通過上述的高純度曲克蘆丁的制備方法制得。
相對于現有技術,本發明包括以下有益效果:本發明在羥乙基化(醚化)反應的某一特定節點上穿插進行特定的絡合反應,將蘆丁分子中裸露的5-OH和4-=O基團先行掩蔽,然后再作后段醚化反應。至反應完全后加酸水解,以脫除保護基團。水解反應液經濃縮并經精制后能夠輕松得到純度非常高的注射用曲克蘆丁。
本發明提供的制備方法可使三羥乙基蘆丁的轉化率得以最大化,從而使原料蘆丁得到最大程度的利用,而且后續分離過程簡便、高效,尤適合注射級高純曲克蘆丁原料藥的工業化生產。
另外,按照上述條件制備三羥乙基蘆丁含量為95~98%的曲克蘆丁的千克產品,原、輔料成本加和后為350~400元;若制備三羥乙基蘆丁含量在98%以上的高純曲克蘆丁,則每千克產品原料成本為420~460元,原料成本都較低。而且其無需采用繁復的離子交換或大孔樹脂脫鹽純化過程,生產周期也會大幅縮短。在本發明技術條件下,車間生產成本可以控制為每千克產品600~750元,若生產色譜純度為90~95%的注射級曲克蘆丁,則只需經過一次結晶即可充分滿足質量要求,車間生產成本將低于每千克500元。
附圖說明
為了更清楚的說明本發明實施例或現有技術中的技術方案,下面將對實施例或現有技術描述中所需要使用的附圖作簡單的介紹,顯而易見地,下面描述中的附圖僅僅是本發明的一些實施例,對于本領域普通技術人員來講,在不付出創造性勞動的前提下,還可以根據這些附圖獲得其他的附圖。
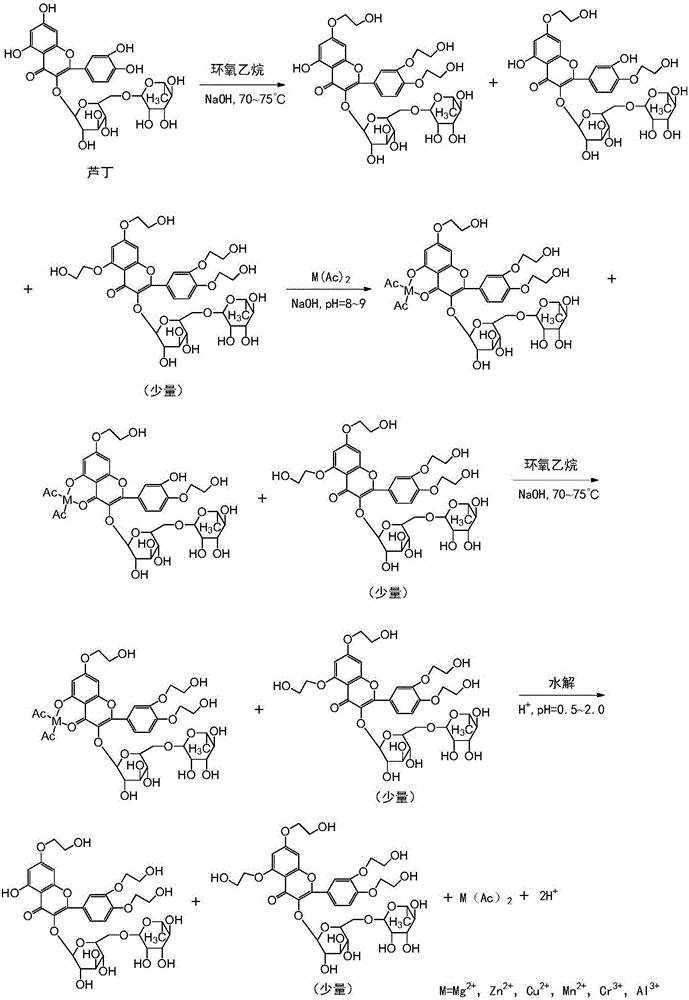
圖1是本發明提供的高純度曲克蘆丁的合成路線示意圖。
具體實施方式
為使本發明實施例的目的、技術方案和優點更加清楚,下面將對本發明實施例中的技術方案進行清楚、完整地描述。實施例中未注明具體條件者,按照常規條件或制造商建議的條件進行。所用試劑或儀器未注明生產廠商者,均為可以通過市售購買獲得的常規產品。
下面對本發明實施例的高純度曲克蘆丁及其制備方法進行具體說明。
高純度曲克蘆丁的制備方法包括步驟S1——前段醚化步驟:將蘆丁與環氧乙烷在醇和水的混合溶劑系統、堿性催化劑的條件下進行醚化反應,并保持反應液中四羥乙基蘆丁的峰面積百分比在1.5%以下(利用高效液相色譜法檢測),停止前段醚化反應;其中,蘆丁、前段醚化步驟中的所述環氧乙烷、醇、水、堿性催化劑的質量比為1:0.1~0.4:1.5~6.5:0.5~8.0:0.008~0.012,醚化反應的反應溫度為60~80℃。
其中,反應液是指醚化反應過程中的反應液。醚化反應可以按照常規醚化反應操作進行,具體的,醚化反應為:將醇、水的混合溶劑與堿性催化劑混合,往其中通保護氣體10~30min,然后加熱至60~65℃,加入蘆丁,再通入環氧乙烷于60~80℃條件下進行反應,并控制反應液的pH為8.0~9.0;保護氣體選自氮氣、惰性氣體中的一種或多種,優選氮氣。氮氣成本低。一般來說,醚化反應時間為6~10h。
本發明中,蘆丁優選液相純度在96%以上的精制蘆丁,用于制備高純度曲克蘆丁,其雜質含量較少,成本低,性價比高,尤其適用于工業生產。
實際操作時,可以在反應瓶中先加入配比量的水和堿性催化劑,攪拌溶解后再加入醇,通保護氣體10~30min,去除水中的游離氧,避免其對后續反應的不良影響。接著水浴升溫至60~65℃后,加入配比量的精制蘆丁,同時緩緩通入環氧乙烷,于60~80℃、pH為8.0~9.0的條件下持續攪拌反應,至反應液完全透明后,每隔0.8~1.2h取樣(利用高效液相色譜法)作三羥乙基蘆丁和異構體的液相檢測,至四羥乙基蘆丁峰面積接近或達到1.5%時立即停止通環氧乙烷。
其中,醇選自甲醇、乙醇、異丙醇中的一種或多種,優選甲醇;水選自純化水、去離子水、超純水中的一種或多種,優選純化水,純化水在醫藥行業中應用較廣;堿性催化劑選自氫氧化鈉、氫氧化鉀中的一種或多種。
高純度曲克蘆丁的制備方法還包括步驟S2——絡合步驟:調節反應溫度為70~90℃(優選為75~85℃),然后在保護氣體的保護下往反應液中加入飽和的醋酸鹽的水溶液,并在加入期間控制反應液的pH為8.0~9.5(優選滴加稀堿水來控制pH),加畢,攪拌10~20min;使之與醚化產物中的三羥乙基蘆丁(也包括少量的二羥乙基,乃至更少量的一羥乙基蘆丁和四羥乙基蘆丁)分子中的4-位羰基和5-位羥基充分發生絡合反應;其中,保護氣體選自氮氣、惰性氣體中的一種或多種,醋酸鹽為金屬離子醋酸鹽或金屬離子醋酸鹽的水合物,蘆丁與醋酸鹽的摩爾比為1:1.0~1.3。
醋酸鹽選自醋酸鎂、醋酸鋅、醋酸銅、醋酸錳、醋酸鉻、醋酸鎳、醋酸鈷、醋酸鎘、醋酸鑭、醋酸釹、醋酸釤中的一種或多種,優選醋酸鎂或醋酸鋅或醋酸鉻,也可以選用上述各種醋酸鹽的水合物。
分散在蘆丁分子A、B和C環的7,3′,4′和5-OH四個羥基的pKa值既有所不同,又相差不大。因此,當與環氧乙烷發生醚化反應時,其優先順序是7-OH,3′-OH,4′-OH和5-OH。由于4′-OH和5-OH的pKa值最為接近,因此,當輪到4′-OH開始醚化時,5-OH的醚化反應緊隨其后。換言之,當三羥乙基蘆丁吸收峰面積接近最大值時,四羥乙基蘆丁的吸收峰已經出現,然后二者幾乎同步增長。因此,為使三羥乙基蘆丁最大程度地生成,同時又能充分抑制四羥乙基蘆丁的產生,便是本發明所要解決的最關鍵的技術。
蘆丁的化學結構屬于黃酮醇苷,其母核由3個環構成,大多數含有羥基或羰基。此結構具有超離域度,整個分子形成一個大π鍵共軛體系,具有強烈的螯合作用,其中的氧原子具有強配位能力,能與許多金屬離子形成穩定的五元環或六元環配位化合物。這些金屬離子包括但不限于Mg2+、Zn2+、Cu2+、Mn2+、Fe2+、Cr3+和Al3+。本發明的發明人通過對比試驗后發現上述2價或3價金屬離子與5-OH、4-=O絡合能力比B3+更強,更何況硼砂為有毒物,作為藥物合成助劑,顯然是不可取的。本發明優選醋酸鎂、醋酸鋅和醋酸鉻作為螯合劑,蘆丁鎂、蘆丁鋅、蘆丁鉻的絡合物結構式分別如下:
蘆丁鎂、蘆丁鋅和蘆丁鉻的配合物的化學計量比都是1:1,而且優先與5-OH,4-=O基團絡合,恰與目標一致。
本發明的另一設計理由是蘆丁A環上5-OH與B環4-=O與上述2價或三價金屬鹽溶液所形成的絡合物在酸性條件下可以使金屬離子解離出來,從而實現本發明制備高純度的曲克蘆丁的目的。
本發明的示例合成路線如圖1所示。其中,M=Mg2+、Zn2+、Cu2+、Mn2+、Cr3+、Al3+。為了如實反映中段絡合反應及其前后段醚化反應化學過程,特將前段醚化反應的副產物、絡合反應后的副產物以及水解后的副產物(少量)均在合成路線圖中標示。
高純度曲克蘆丁的制備方法還包括步驟S3——后段醚化步驟:調節反應溫度為60~80℃、反應液的pH為8.0~9.0,繼續通入環氧乙烷進行反應,同時利用高效液相色譜法對反應液進行在線檢測,至三羥乙基蘆丁的峰面積不再增長(即達到最大值)時,終止醚化反應,得到反應產物;其中,蘆丁與后續醚化步驟中的環氧乙烷的質量比為1:0.1~0.3。一般來說,后段醚化步驟中,反應時長為1~5h。
此步驟中寫到的是“終止醚化反應”,是因為本發明中,實際上醚化反應是分成了兩個階段,一個是前段醚化,一個是后段醚化,前段醚化反應與后段醚化反應的反應溫度相同、pH也相同。
由于反應體系中可能殘存有過量環氧乙烷,所以在后段醚化步驟之后,在后處理步驟之前,還包括:將反應產物降至5~25℃,同時往反應產物中通入保護氣體,排除未作用的環氧乙烷,保護氣體選自氮氣、惰性氣體中的一種或多種,優選氮氣。
高純度曲克蘆丁的制備方法還包括步驟S4——后處理步驟:利用稀酸與反應產物發生水解反應,然后濃縮、精制。
其中,稀酸選自質量濃度為5~15%的稀硫酸、摩爾濃度為2~6mol/L的稀鹽酸中的一種或多種,水解反應是在保護氣體條件下進行,具體操作為:利用稀酸將反應產物酸化至pH在1.5以下,攪拌10~20min后,在氮氣流氛圍下利用質量濃度為1~15%的稀堿溶液回調pH至2.5~3.5。其中,保護氣體依然選自氮氣、惰性氣體中的一種或多種,優選氮氣。綜合考慮稀酸的酸性及水解后生成的金屬鹽在醇溶劑中的溶解性,在大多數情況下,此稀酸優選質量濃度5.0~15.0%的稀硫酸,少數情況下選擇摩爾濃度為2.0~6.0mol/L的稀鹽酸。利用稀酸水解脫除保護基團后,用于回調的稀堿溶液可以選用質量濃度為1.0~5.0%的氫氧化鈉或氫氧化鉀水溶液,質量濃度為1.0~10.0%的氨水,質量濃度為5.0~10.0%的碳酸鈉水溶液,質量濃度為10.0~15.0%的碳酸氫鈉或碳酸氫銨水溶液,優選碳酸鈉溶液,回調pH值能夠提高反應產物體系的穩定性,同時可降低酸根離子對未來金屬反應設備的腐蝕性。
而后,濃縮、精制水解反應的產物包括:將水解產物在60~80℃水浴上真空(真空度≥650mmHg)濃縮至含水量在25%以下(優選在20%以下),得到濃縮物,然后利用鎂、鋅、鉻的硫酸鹽不溶于醇的性質,用醇溶液對濃縮物精制2~3次,醇溶液選自甲醇、乙醇中的任意一種,濃縮物與醇溶液的質量比為1:3.5~8.5。若醇溶液為甲醇,則濃縮物與醇溶液的質量比為1:3.5~6.5(優選質量比為1:4.5~5.5),若醇溶液為乙醇,因蘆丁在乙醇中溶解性較小,所以醇溶液用量需相應增大。
利用醇溶液對濃縮物精制具體為:將醇溶液與濃縮物混合后加熱溶解,并加入活性炭回流脫色,濃縮物與活性炭的質量比為1:0.01~0.05,再在5~15℃下結晶2~8h(優選3~6h),過濾。
實際操作時,可以將醇溶液加入濃縮物中,然后水浴加熱,加熱溫度即為醇溶液的沸點溫度,全溶后加入活性炭回流脫色10~30min,而后過濾,使水解下來的金屬鹽和部分難溶于醇溶液的非蘆丁成分以及炭渣被截留。將過濾后的濾液于5~15℃冷卻結晶,再次過濾,得到三羥乙基蘆丁液相純度≥96.0%的曲克蘆丁(一次精制品)。
精制還包括:一次精制品用含水甲醇回流溶解后再冷卻結晶、過濾、真空干燥后得到三羥乙基蘆丁液相純度≥98%的曲克蘆丁二次精制品。在二次結晶過程中,一次精制品與甲醇的質量比為1:4.0~8.0;含水甲醇的濃度為90.0~98.0%,冷卻結晶溫度為5.0-12.0℃。二次精制品經用甲醇熱洗一次,再冷卻沉淀、過濾、干燥后得到三羥乙基蘆丁液相純度≥99%的更高純度曲克蘆丁(也稱作7,3’,4’-三羥乙基蘆丁單體)。甲醇熱洗過程中二次精制品與甲醇的質量比為1:5.0~10.0,洗滌溫度為50℃回流溫度,熱洗時間不少于10min,沉降溫度5~10.0℃,真空干燥溫度:50~65℃。
本發明還提供了一種高純度曲克蘆丁,是通過上述的高純度曲克蘆丁的制備方法制得。該高純度曲克蘆丁中,7,3’,4’-三羥乙基蘆丁含量在98%以上。
以下結合實施例對本發明的特征和性能作進一步的詳細描述:
實施例一
本實施例提供的高純度曲克蘆丁的制備方法,包括以下步驟:
S1:醚化反應為:將異丙醇、無鹽水和氫氧化鉀混合,往其中通氬氣10min,然后加熱至60~63℃,加入液相純度為96.10%的精制蘆丁,再通入環氧乙烷,并控制反應pH為8.0~8.3、反應溫度60~65℃下持續進行醚化反應,直至通過高效液相色譜法檢測出反應液中四羥乙基蘆丁峰面積比為0.55%時停止通氣反應;其中,蘆丁、環氧乙烷、異丙醇、無鹽水、氫氧化鉀的質量比為1:0.32:1.5:2.0:0.008;
S2:調節物質(反應液)的pH為8.0~8.5、溫度為70~75℃,往反應液中加入醋酸錳,攪拌13min,蘆丁與醋酸錳的摩爾比為1:1.0;
S3:調節反應液的溫度為60~65℃,往反應液中通入環氧乙烷維持反應液的pH為8.0~8.3,在該條件下繼續反應,同時利用高效液相色譜法對三羥乙基蘆丁進行在線液相檢測,至物質中三羥乙基蘆丁的峰面積(93.16%)不再增長時,停止反應,得到反應產物;
S4:在氮氣通入的條件下,滴加質量濃度為10%的稀硫酸將反應產物酸化至pH為0.5~0.6,加酸完畢,繼續攪拌水解12min后利用質量濃度為8%的碳酸鈉回調pH至2.8,在真空條件下將水解產物濃縮至含水量為15.2%,得到濃縮物;
S5:一次結晶:將乙醇與濃縮物粗品混合并加熱溶解,加入活性炭回流脫色15min,過濾除炭渣和不溶性金屬鹽和活性炭渣,濾液冷卻至5~6℃攪拌結晶3h,經過濾、真空干燥后得到曲克蘆丁一次精制品;其中,縮物(折干計)、乙醇、活性炭的質量比為1:4.5:0.01;
S6:二次結晶:將一次結晶物與濃度為90.0%的甲醇混合后加熱至沸,并回流15min,然后冷卻至5~7℃攪拌結晶3.5h,過濾后于50~60℃真空干燥;
S7:洗滌:將二次結晶品與配量的甲醇混合,并于55~60℃條件下攪拌洗滌15min,冷卻至6~7℃,陳化3.0h,過濾、于55~60℃真空干燥至干,得到高純曲克蘆丁。
實施例二
本實施例提供的高純度曲克蘆丁的制備方法,包括以下步驟:
S1:將乙醇、超純水和氫氧化鈉混合,往其中通氮氣12min,然后加熱至65℃,加入液相純度為97%的精制蘆丁,再通入環氧乙烷,控制反應液的pH為8.7~9.0、反應溫度為75~80℃,持續進行醚化反應,至反應液澄清后,前期每隔1.2h取樣做PHLC檢測,至反應液中四羥乙基蘆丁峰面積為0.8%時停止通氣反應;其中,蘆丁、環氧乙烷、乙醇、超純水、氫氧化鈉的質量比為1:0.32:2.5:2.5:0.012;
S2:調節反應液的pH為9.2~9.5、溫度為86~90℃,往反應液中滴加配比量的醋酸鉻溶液,加畢,繼續攪拌18min,蘆丁與醋酸鉻的摩爾比為1:1.3;
S3:調節反應液的溫度為78~80℃,往反應液中通入環氧乙烷,并控制反應液的pH為8.7~9.0,繼續進行醚化反應,同時利用HPLC法作在線液相檢測,至反應液中三羥乙基蘆丁的峰面積不再增長時,停止反應,得到反應產物;
將反應產物降至25℃,同時往反應產物中通入氦氣,排除未作用的環氧乙烷;
S4:在氬氣的環境條件下,利用質量濃度為15%的稀硫酸將反應產物酸化至pH為1.45,加酸完畢,繼續攪拌水解17min后利用質量濃度為10%的碳酸鈉溶液回調pH至2.5,在真空條件下將水解產物濃縮至含水量為24.5%,得到濃縮物;
S5:將甲醇與濃縮物混合后加熱溶解,再加入活性炭回流脫色30min,真空過濾除去炭渣和不溶性金屬鹽,濾液冷卻至13~15℃,靜置結晶8h后過濾,濾餅于55~60℃真空干燥至干,得到曲克蘆丁一次結晶品;其中,濃縮物、甲醇、活性炭的質量比為1:5.5:0.05;
S6:二次結晶:將一次結晶物與濃度為97.5%甲醇混合后加熱至沸,并回流30min,冷卻至11~12℃攪拌結晶6.5h,過濾后50~60℃真空干燥;
S7:洗滌:將二次結晶品與濃度為99.0%甲醇混合,于回流條件下攪拌洗滌25min,冷卻至9~10℃,陳化6.0h,過濾、于55~60℃真空干燥至干,得到高純曲克蘆丁。
實施例三
本實施例提供的高純度曲克蘆丁的制備方法,包括以下步驟:
S1:于3000mL并配備梅特勒酸度-溫度計、導氣管、出氣管、冷凝器和電動攪拌機的反應瓶中依次投入350mL純化水和3.96g氫氧化鈉,全溶后再加入350mL甲醇,通氮氣15min后一次加入液相純度為96.2%的精制蘆丁317.5g(0.5mol),水浴升溫至65℃時停止通入氮氣,通過導氣管向反應瓶中緩緩通入環氧乙烷氣體進行前段醚化反應。期間,維持內溫65~75℃、pH為8.3~8.6的條件持續通氣反應,直至反應液變清。然后定時取樣做三羥乙基蘆丁及其三個羥乙基異構體的液相純度檢測。當四羥乙基蘆丁峰面積比達到1.15%時,立即終止醚化反應。同時用氮氣驅趕反應瓶內剩余環氧乙烷氣體。
S2:繼續通氮氣,并控制內溫為75~85℃,間歇滴加5.0%氫氧化鈉溶液調節反應液的PH為8.3~8.8,自滴液漏斗中向反應瓶中均勻滴加質量濃度為20%的四水醋酸鎂(118g,0.55mol)水溶液,使與蘆丁羥乙基化物發生螯合反應。加畢,維持反應液的pH為8.3~8.8,繼續攪拌反應15min。
S3:螯合反應結束后,降溫至70℃,并于70~75℃、反應液pH為8.3~8.8的條件下繼續通入環氧乙烷進行后段醚化反應,并定時取樣測定反應液中三羥乙基蘆丁液相純度。直至三羥乙基蘆丁峰面積百分比達到93.3%,并且不再上升時立即停止后段醚化反應。同時用水浴降溫,并通氮氣充分驅趕殘存的環氧乙烷余氣。
S4:當反應液內溫降至25℃時,于攪拌下滴加質量濃度為12.5%稀硫酸酸化至PH為1.05,加畢,繼續攪拌反應15min。然后滴加質量濃度為10%的碳酸鈉水溶液,將反應液PH值回調至3.0。
S5:將全部反應液轉入濃縮瓶中,于水浴溫度70-80℃條件下真空旋轉濃縮至含水量為15.0%時加入2350mL甲醇并繼續加熱旋轉使溶解。加入3.5g活性炭,回流脫色30min后趁熱過濾。再將濾液放冷至40℃,靜置2h后作真空過濾,并棄去不溶性過濾物。
S6:將上述濾液緩慢冷卻至6℃,慢速攪拌結晶4h后真空吸濾,并用少量冷溶媒洗滌濾餅3次。濾餅于60℃真空干燥,得301g曲克蘆丁一次精制品。經液相檢測,三羥乙基蘆丁液相純度為96.26%,含量96.11%,其他指標檢測結果列于表1。以三羥乙基蘆丁計,醚化反應、一次精制過程總摩爾收率為80.73%。
S7:二次精制:取一次精制品250g于3000mL反應瓶中,加入體積濃度為95%的含水甲醇2100mL,攪拌下水浴升溫至沸,回流20min后趁熱過濾,棄除少量不容物后將濾液轉入結晶瓶中,常溫水攪拌降溫至35℃時再依次換用涼水、冰水繼續緩慢攪拌降溫至7℃,再慢速攪拌結晶4h后吸濾,并用少量冷溶媒洗滌濾餅3次,濾餅于60℃真空干燥,得232.5g曲克蘆丁二次精制品,液相檢測三羥乙基蘆丁液相純度為:98.37%(曲克蘆丁含量98.23%),其他指標檢測數據見表1。以三羥乙基蘆丁計,二次精制總摩爾收率為92.73%。
S8:洗滌(三次精制):取二次精制品200g于2000mL反應瓶中,加入1600mL的甲醇。攪拌下水浴升溫至沸,攪拌回流洗滌15min。洗畢,先自然降溫,再用冷水和冰水降溫至9℃,靜置4h后過濾。濾餅經用新鮮甲醇洗滌2次。濾餅于60~62℃真空干燥至干,得三次精制品190.3g。三羥乙基蘆丁液相純度99.41%,曲克蘆丁含量:99.30%。其他分析數據見附表1。三次精制收率為94.5%。醚化反應、三次精制過程總摩爾收率為70.74%。
表1一~三次精制產品主要質量控制項目檢測結果匯總
實施例四
本實施例是在實施例三的投料規模和工藝條件基礎上,以二水醋酸鋅代替四水醋酸鎂作為螯合劑。精制蘆丁與二水醋酸鋅的摩爾比為1:1.1,二水醋酸鋅水溶液的配置質量濃度為20.0%、加料方式、絡合反應PH值、溫度控制范圍、后續酸化過程使用稀硫酸質量濃度、酸化PH值、回調PH過程使用堿水濃度、回調終點PH值以及其他條件均與實施例三相同。本實施例的醚化反應、水解、濃縮和一、二、三次精制過程工藝條件與實施例三相同。以三羥乙基蘆丁計,醚化反應、一次精制過程總摩爾收率為81.57%,二次精制總摩爾收率為92.46%,三次精制收率為94.73%。醚化反應、三次精制過程總摩爾收率為71.45%。三次精制產品品各項檢測數據列于表2。
表2一~三次精制產品主要質量控制檢測結果匯總
對照例一
本實施例中醚化反應一氣呵成,直至終點(中間階段不設絡合反應步驟)。其中,反應過程中原料精制蘆丁、氫氧化鈉、純化水和甲醇投料量與實施例三相同。環氧乙烷通入流量以反應液PH值處于8.0~9.0范圍內為準,用量以實際耗用量計。醚化反應至三羥乙基蘆丁面積百分比達到77.74%,且不再上升時終止反應。依法冷卻、通氮氣驅趕剩余的環氧乙烷。然后用濃度為6mol/L的稀鹽酸直接酸化至PH為3.0~3.5。酸化以后的真空濃縮和三次精制過程與物料配比與實施例三、實施例四完全一致。制備過程總摩爾收率61.52%。各精制階段產品檢測數據列于表3。
表3一~三次精制產品主要質量控制檢測結果匯總
對照例二
本實施例提供的制備方法,是仿制CN103113437A的實施例一而做出的:53g藥用蘆丁(含量:94.05%)分散在150mL純化水中,加入0.6g氫氧化鈉,在攪拌均勻后加入環氧乙烷12g,73℃反應3.5h。取終點反應液做液相檢測,一羥乙基蘆丁2.86%,四羥乙基蘆丁5.27%,二羥乙基蘆丁16.56%,三羥乙基蘆丁73.74%,二、三羥乙基蘆的峰面積總和90.30%。然后加入硼砂31g,測定反應液PH為9.36,加入環氧乙烷4.1g,繼續在70℃反應2.5h。再取終點反應液做液相監測,曲克蘆丁峰面積85.29%,四羥乙基蘆丁10.36%,二羥乙基蘆丁1.92%。然后滴加濃鹽酸中和PH至5.87。將中和后的液體減壓濃縮至干,加入200mL體積濃度為90%的乙醇,然后繼續用6mol/L的稀鹽酸調節PH至3.06,加熱至74~76℃持續攪拌25min,稍冷后真空過濾,除去不溶物后將濾液緩慢降至室溫,靜置4h后吸濾。濾餅經干燥后,得到黃色粉末35.6g。總摩爾收率:54.72%。產品分析結果見表4。
表4一次精制產品主要質量控制檢測結果匯總
以上所述僅為本發明的優選實施例而已,并不用于限制本發明,對于本領域的技術人員來說,本發明可以有各種更改和變化。凡在本發明的精神和原則之內,所作的任何修改、等同替換、改進等,均應包含在本發明的保護范圍之內。
- 還沒有人留言評論。精彩留言會獲得點贊!