一種具有擠壓和開關(guān)效應(yīng)的納米藥物載體的制備方法與流程
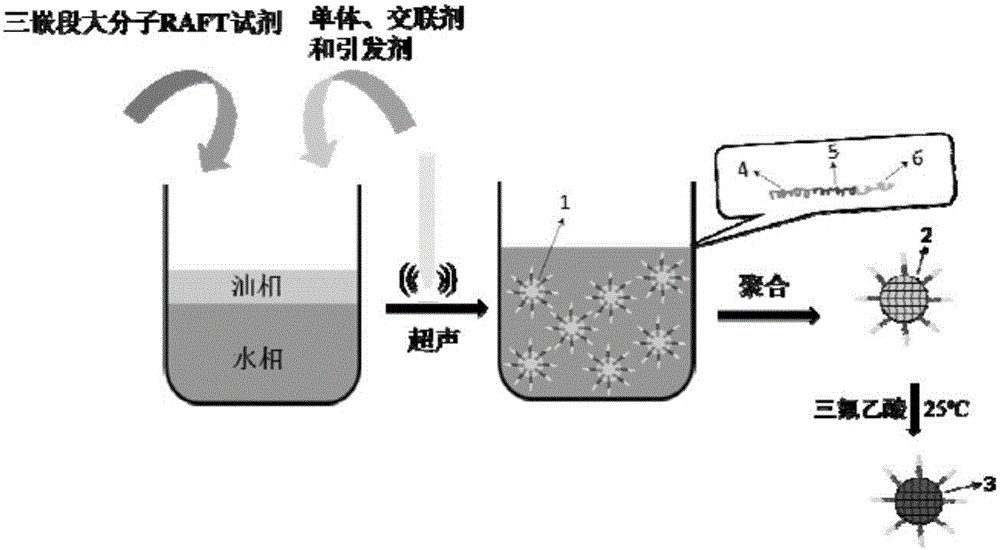
本發(fā)明涉及一種具有擠壓和開關(guān)效應(yīng)的納米藥物載體的制備方法,利用該法可以制備得到一種具有核殼結(jié)構(gòu)的高分子納米球,進一步通過靜電作用載藥后可以實現(xiàn)在酸性環(huán)境下的快速釋放,是一種優(yōu)良的藥物定向釋放載體。屬于生物醫(yī)用高分子材料領(lǐng)域。
背景技術(shù):
與傳統(tǒng)藥物制劑相比,納米藥物載體具有延長藥物在體內(nèi)的半衰期、提高藥物生物利用率和有效血藥濃度、包埋疏水性藥物、降低用藥次數(shù)、實現(xiàn)靶向給藥(靶向基團修飾后)等特點,在疾病治療特別是癌癥化療方面具有無可比擬的優(yōu)勢(Advanced Drug Delivery Reviews,2002,54:631-651;Nature Materials,2013,12:991-1003)。截至目前科研人員已經(jīng)開發(fā)制備了多種多樣的納米藥物載體,包括膠束、微囊、脂質(zhì)體、樹狀大分子、聚合物-藥物偶聯(lián)物以及無機納米球等(Langmuir,2014,30:9819-9827)。刺激響應(yīng)型納米藥物載體能夠通過感應(yīng)外部環(huán)境的變化(溫度、pH、光、磁場、離子強度等)或在酶的作用下實現(xiàn)藥物的釋放,在實現(xiàn)藥物在體內(nèi)的定向釋放方面具有很大的潛力。
我們知道人體消化道內(nèi)存在天然的pH梯度(pH1-7.5),此外感染/炎癥部位(pH5.4-5.7)、腫瘤部位(pH5.8-7.2)、細(xì)胞內(nèi)涵體和溶酶體(pH4.5-5.5)等特定部位的pH都低于正常體液(pH7.4)。這些差別使得pH響應(yīng)型納米藥物載體的研究開發(fā)成為熱點。人們可以在器官、組織以及細(xì)胞器水平上設(shè)計多種pH響應(yīng)型納米載體(Molecular Pharmaceuticals,2010,7:1913-1920)。pH響應(yīng)型納米藥物載體的釋藥機理通常有以下四種:1)藥物通過酸敏感共價鍵偶聯(lián)到載體上,在酸性條件下實現(xiàn)藥物釋放;2)離子聚合物構(gòu)成的殼層/殼層通過在酸性條件下溶脹增加藥物釋放速率;3)通過離子聚合物在酸性條件下破壞膠束/脂質(zhì)體的結(jié)構(gòu)實現(xiàn)藥物釋放;4)兩性離子聚合物在酸性條件下實現(xiàn)載體表面電荷由負(fù)電到正電的轉(zhuǎn)變,從而增加細(xì)胞內(nèi)吞量。要想實現(xiàn)pH響應(yīng)型藥物載體在pH7時具有高載藥量和低泄漏率,在pH5-6時能夠快速可控釋放藥物是比較困難的。并目前文獻報道的大多數(shù)pH響應(yīng)型藥物載體往往合成步驟復(fù)雜(多步反應(yīng)偶聯(lián)藥物或載藥后多步純化)。大多數(shù)給藥系統(tǒng)的藥物釋放都是以濃度差為驅(qū)動力的擴散(菲克第一定律),盡管能做到快速響應(yīng),很難做到藥物的快速釋放。
具有擠壓功能的pH響應(yīng)型給藥系統(tǒng)可以突破濃度擴散限制,實現(xiàn)藥物快速釋放。Kim等人將裝有藥物的水凝膠置于有兩個釋放小孔的剛性膠囊(0.6ml)中,把膠囊置于釋放介質(zhì)溶液中,水凝膠馬上溶脹堵塞小孔。當(dāng)介質(zhì)溶液的pH值或pH值/溫度改變時,水凝膠開始收縮,打開小孔并從凝膠基質(zhì)中機械擠壓出藥物溶液,排出的藥液通過小孔迅速擴散到裝置外(Journal of Controlled Release,1997,48:141-148)。這種給藥系統(tǒng)被用來運送在胃部易分解的藥物以及對胃有刺激、會在胃腸道上部產(chǎn)生有害作用的藥物,只適合于口服。最近,Bronich等人(Journal of Controlled Release,2009,138:197-204)制備了負(fù)載阿霉素的pH敏感型膠束。他們首先利用Ca2+驅(qū)動商品化的聚乙二醇-聚甲基丙烯酸自組裝,然后利用乙二胺交聯(lián)聚甲基丙烯酸內(nèi)核。這種納米載體在pH7時可以通過靜電作用達(dá)到75%(質(zhì)量比)的載藥率,在pH5.5時通過交聯(lián)內(nèi)核的收縮快速釋放阿霉素。然而這種交聯(lián)膠束在pH7.4時高度溶脹,阿霉素的泄漏比較嚴(yán)重,48h內(nèi)有超過60%的阿霉素被釋放出來,降低了它的臨床應(yīng)用價值。
技術(shù)實現(xiàn)要素:
針對現(xiàn)有pH響應(yīng)型納米藥物載體存在的問題,本發(fā)明采用自制的三嵌段兩親性聚合物作為表面活性劑和微乳液聚合的大分子可逆加成-斷裂鏈轉(zhuǎn)移(RAFT)試劑,通過微乳液活性聚合制備具有核殼結(jié)構(gòu)的納米載體,進一步水解后得到pH響應(yīng)型納米載體(請見附圖1)。這種納米載體可以作為凍干粉儲存,對帶正電的藥物具有很高的負(fù)載率。在pH7.4時通過殼層的開關(guān)作用有效降低了藥物的釋放,在pH5.0時通過機械擠壓作用迅速釋放藥物,后者的藥物釋放速率是前者的3倍以上,是一種在腫瘤化療領(lǐng)域具有很大應(yīng)用潛力的pH響應(yīng)型納米給藥系統(tǒng)。
本發(fā)明所采用的技術(shù)方案是:
一種具有擠壓和開關(guān)效應(yīng)的納米藥物載體的制備方法,所述制備方法包括如下步驟:
(1)以聚乙二醇單甲醚(甲基)丙烯酸酯為單體,采用可逆加成-斷裂鏈轉(zhuǎn)移聚合(RAFT)方法制備線性聚合物,反應(yīng)溫度為55-85℃;反應(yīng)時間為3-24h;單體濃度0.1-5M,單體和RAFT試劑的摩爾比范圍為10:1-100:1,RAFT試劑與引發(fā)劑的摩爾比范圍為1:0.05-1:0.2,反應(yīng)溶劑可選甲苯、乙苯、氯苯、二氧六環(huán)、二甲基甲酰胺、二甲基乙酰胺、苯甲醚、乙醇、異丙醇等有機溶劑的一種;內(nèi)標(biāo)物可選均三甲苯、三惡烷等。反應(yīng)后的產(chǎn)品在正己烷、環(huán)己烷或乙醚中沉淀兩次,離心后,所得高分子真空干燥后進入步驟(2);
(2)以步驟(1)所得的第一嵌段線性聚合物作為大分子RAFT試劑,選用陽離子單體作為第二嵌段單體,采用RAFT聚合得到兩嵌段聚合物。反應(yīng)后的產(chǎn)品在正己烷或環(huán)己烷中沉淀兩次,其他反應(yīng)條件同步驟(1);
(3)以步驟(2)所得的嵌段共聚物作為大分子RAFT試劑,選用(甲基)丙烯酸叔丁酯作為第三嵌段單體,采用RAFT聚合得到三嵌段聚合物。反應(yīng)溶劑可選甲苯、乙苯、氯苯、苯甲醚、二氧六環(huán)等有機溶劑的一種,反應(yīng)后的產(chǎn)品在正己烷或環(huán)己烷中沉淀兩次,其他條件同步驟(1);
(4)將步驟(3)所得的三嵌段兩親性聚合物溶于去離子水中配成水相;(甲基)丙烯酸叔丁酯、交聯(lián)劑、引發(fā)劑和內(nèi)標(biāo)物均三甲苯混合配成油相,交聯(lián)度(交聯(lián)劑占單體和交聯(lián)劑質(zhì)量和的百分?jǐn)?shù))范圍為5-50%;油相加入水相后利用超聲波破碎儀在冰水浴中制備微乳液。所得微乳液轉(zhuǎn)入帶有橡皮塞的單口瓶,充氮氣除氧30min后升溫開始RAFT聚合,反應(yīng)溫度為55-85℃,反應(yīng)一段時間后得到具有核殼結(jié)構(gòu)的聚(甲基)丙烯酸叔丁酯納米球。將所得的納米球溶液用透析袋透析24h,透析介質(zhì)依次為無水乙醇和去離子水,目的是除去未參加反應(yīng)的物質(zhì),然后將透析袋里的納米球溶液用真空凍干機干燥,得到聚(甲基)丙烯酸叔丁酯納米球凍干粉。
(5)將步驟(4)所得的產(chǎn)品加入到90%(體積比)的三氟乙酸水溶液中,室溫下反應(yīng)過夜脫除叔丁基保護,然后將反應(yīng)混合物在去離子水中透析24h,將透析袋里的納米球溶液用真空凍干機干燥,得到具有擠壓和開關(guān)效應(yīng)的pH響應(yīng)型聚(甲基)丙烯酸納米球干粉。
(6)將步驟(5)所得的納米球干粉分散在藥物的pH緩沖溶液中,藥物的濃度范圍10-2000μg/ml,pH緩沖液pH值范圍6-8。藥物分子通過靜電作用吸附到載體上,室溫下混合24h后通過離心分離得到載藥納米球。
優(yōu)選的,步驟(1)-(4)中的引發(fā)劑為常規(guī)油溶性自由基聚合引發(fā)劑,選自偶氮二異丁腈、偶氮二異庚腈、偶氮二異丁酸二甲酯、偶氮二環(huán)己基甲腈、過氧化苯甲酰、過氧化苯甲酰叔丁酯、過氧化甲乙酮等中的一種。反應(yīng)溫度為60-70℃;反應(yīng)時間為5-21h。步驟(1)-(3)中單體濃度為1-3M。
步驟(1)-(4)中單體的轉(zhuǎn)化率通過核磁分析反應(yīng)前后的反應(yīng)液可以確定。
步驟(1)中所述的RAFT試劑為4-氰基-4-(苯基硫代甲酰硫基)戊酸(CPADB)、4-氰基-4-(丙基三硫代碳酸酯基)戊酸(CPPT)、4-氰基-4-(十二烷基三硫代碳酸酯基)戊酸(CDPT)二硫代苯甲酸枯酯(CDB)、2-氰基-2-丙基苯并二硫(CPDB)等常見RAFT試劑的一種。
陽離子單體是指在酸性條件下可以質(zhì)子化的單體,優(yōu)選的,步驟(2)中所述的陽離子單體為(甲基)丙烯酸N,N-二甲氨基乙酯和(甲基)丙烯酸N,N-二乙氨基乙酯這四種單體中的至少一種,但是并不局限于以上四種。
步驟(2)中所述大分子RAFT試劑的分子量范圍1000-30000Da,優(yōu)選5000-15000Da。
步驟(2)中陽離子聚合物分子量的控制很關(guān)鍵,它主要起到納米藥物載體的開關(guān)作用,過低起不到開關(guān)作用,過高會增加藥物在酸性條件下的擴散阻力。分子量的控制通過反應(yīng)時間、單體濃度以及單體與RAFT試劑的摩爾比來控制。
步驟(3)中所述大分子RAFT試劑的分子量范圍1500-40000Da,優(yōu)選5000-20000Da。
步驟(3)中第三嵌段聚合物的分子量大小也很關(guān)鍵,它直接影響著步驟(4)中水包油型微乳液的穩(wěn)定性。分子量的控制方法與步驟(2)相同。
步驟(3)中所得的雙親性三嵌段大分子RAFT試劑的親油親水平衡值(HLB),采用Griffin’s方法計算,屬于比值法,公式如下:
其中,M1,M2,M3分別是第一嵌段、第二嵌段和第三嵌段的摩爾質(zhì)量,MH是親水的第一嵌段和第二嵌段摩爾分子量之和,M為三嵌段聚合物分子的總摩爾質(zhì)量。
步驟(4)中所述的交聯(lián)劑為二(甲基)丙烯酸乙二醇酯、二(甲基)丙烯酸丙二醇酯、二(甲基)丙烯酸1,4-丁二醇酯、新戊二醇二(甲基)丙烯酸酯、雙(2-甲基丙烯酰氧乙基)二硫醚中的至少一種;三嵌段聚合物的分子量范圍1800-80000Da;HLB范圍10-18;三嵌段聚合物RAFT穩(wěn)定劑的用量為去離子水質(zhì)量的0.1-2.0%;油相質(zhì)量占水相質(zhì)量的1-20%。
優(yōu)選的,步驟(4)中所述的三嵌段兩親性聚合物分子量范圍5000-20000Da,HLB范圍12-16,加入量為去離子水質(zhì)量的0.3-1.0%;油相質(zhì)量占水相質(zhì)量的5-15%,交聯(lián)度為10-30%。
步驟(4)中超聲波破碎儀的工作振幅和時間會影響納米藥物載體粒徑的大小,可根據(jù)所需粒徑調(diào)節(jié)。超聲破碎破碎儀的振幅范圍30-70%,超聲時間范圍5-30min。
步驟(6)中所述的藥物為在pH6-8的范圍內(nèi)帶正電的藥物,如阿霉素、吉西他濱、表柔比星等。載藥時納米球與藥物的質(zhì)量比范圍1:1-20:1,優(yōu)選1.5:1-10:1。
本發(fā)明所制備的具有擠壓和開關(guān)效應(yīng)的pH相應(yīng)型納米藥物載體的粒徑為80-300nm,具有核殼結(jié)構(gòu)。
本發(fā)明產(chǎn)生的技術(shù)效果是:
本發(fā)明利用RAFT微乳液聚合技術(shù)制備了一種具有核殼結(jié)構(gòu)的納米粒子,進一步水解后得到具有擠壓和開關(guān)功能的pH響應(yīng)型納米藥物載體。該納米載體核層和殼層都具有獨特的功能。核層由交聯(lián)的聚丙烯酸或聚甲基丙烯酸組成,殼層由兩嵌段的聚乙二醇和陽離子聚合物組成。核層在pH值呈酸性的腫瘤或發(fā)炎部位會由溶脹狀態(tài)迅速收縮,從而將包埋的藥物迅速“擠壓”出來;殼層的聚乙二醇片段會使納米粒子在體內(nèi)循環(huán)過程中“隱形”,延長了藥物載體在體內(nèi)的循環(huán)半衰期;殼層的陽離子聚合物片段在中性pH值時會通過靜電作用吸附于核層上,阻止藥物釋放出來,在酸性pH值時靜電作用消失而呈伸展?fàn)顟B(tài),有助于藥物的釋放,起到了“開關(guān)”作用(請見附圖6)。該納米藥物載體在藥物可控釋放領(lǐng)域特別是惡性腫瘤的化療方面具有很大應(yīng)用潛力。
附圖說明:
圖1具有擠壓和開關(guān)效應(yīng)的pH響應(yīng)型納米藥物載體制備過程示意圖;
圖中標(biāo)號1為以三嵌段大分子RAFT為穩(wěn)定劑的微乳液;
標(biāo)號2為具有核殼結(jié)構(gòu)的聚(甲基)丙烯酸叔丁酯納米球結(jié)構(gòu)示意圖;
標(biāo)號3為具有核殼結(jié)構(gòu)的聚(甲基)丙烯酸納米球結(jié)構(gòu)示意圖;
標(biāo)號4為三嵌段聚合物的聚(甲基)丙烯酸叔丁酯片段;
標(biāo)號5為三嵌段聚合物的陽離子聚合物片段;
標(biāo)號6為三嵌段聚合物的聚乙二醇片段。
圖2為本發(fā)明實例1中pH響應(yīng)型納米球水解前后的透射電鏡圖;
圖中a圖為水解前的聚甲基丙烯酸叔丁酯納米球;
b圖為水解后的聚甲基丙烯酸納米球。
圖3為本發(fā)明實例1中pH響應(yīng)型納米藥球水解前后的紅外光譜圖;
圖中a線為水解前的紅外光譜圖;
b線為水解后的紅外光譜。
圖4為本發(fā)明實例1中pH響應(yīng)型納米球水解后粒徑及zeta電勢隨pH的變化圖;
圖中a線為粒徑隨pH值的變化圖;
b線為zeta電勢隨pH值的變化圖。
圖5為本發(fā)明實驗例中pH響應(yīng)型納米球在pH7.4和pH5.0下的藥物釋放曲線圖。
圖中a線為實施例1中載藥納米球在pH5.0下的藥物釋放曲線;
b線為實施例1中載藥納米球在pH7.4下的藥物釋放曲線。
圖6為本發(fā)明中pH響應(yīng)型納米藥物載體“擠壓”和“開關(guān)”功能示意圖。
圖中標(biāo)號1為具有“隱形”功能的聚乙二醇片段;
標(biāo)號2為具有“開關(guān)”功能的陽離子聚合物片段。
具體實施方式
本發(fā)明具體實施方式如下:
實施例1
1)利用RAFT反應(yīng)合成聚乙二醇單甲醚聚甲基丙烯酸酯(PPEGMEMA)
室溫下在50ml單口反應(yīng)瓶中加入攪拌子,然后依次加入偶氮二異丁腈(AIBN)(49.26mg,0.3mmol)、聚乙二醇單甲醚甲基丙烯酸酯(PEGMEMA)(9000mg,30mmol)、4-氰基-4-(苯基硫代甲酰硫基)戊酸(CPADB)(558.8mg,2mmol)、均三甲苯(100μl)及20ml甲苯,翻口橡皮塞密封。待反應(yīng)體系混合均勻后在冰水浴中用針頭通入氮氣除氧20min,將反應(yīng)瓶在65℃油浴鍋中反應(yīng)16h。反應(yīng)結(jié)束后將反應(yīng)液在正己烷中沉淀兩次,離心后真空干燥得到第一嵌段PPEGMEMA。通過核磁氫譜分析單體轉(zhuǎn)化率91.56%,理論分子量4400。
2)利用RAFT反應(yīng)合成聚乙二醇單甲醚聚甲基丙烯酸酯-聚甲基丙烯酸N,N-二甲胺基乙酯(PPEGMEMA-b-PDMAEMA)
室溫下在50ml單口反應(yīng)瓶中加入攪拌子,然后依次加入偶氮二異丁腈(AIBN)(8.21mg,0.05mmol)、PPEGMEMA(2200mg,0.5mmol)、甲基丙烯酸N,N-二甲氨基乙酯(DMAEMA)(3144mg,20mmol)、均三甲苯(100μl)及15ml甲苯,翻口橡皮塞密封。待反應(yīng)體系混合均勻后在冰水浴中用針頭通入氮氣除氧20min,將反應(yīng)瓶在65℃油浴鍋中反應(yīng)15h。反應(yīng)結(jié)束后將反應(yīng)液在正己烷中沉淀兩次,離心后真空干燥得到嵌段共聚物PPEGMEMA-PDMAEMA。通過核磁氫譜分析單體轉(zhuǎn)化率77.93%,理論分子量9300。
3)利用RAFT反應(yīng)合成聚乙二醇單甲醚聚甲基丙烯酸酯-聚甲基丙烯酸N,N-二甲胺基乙酯-聚甲基丙烯酸叔丁酯(PPEGMEMA-b-PDMAEMA-b-PtBMA)
室溫下在50ml單口反應(yīng)瓶中加入攪拌子,然后然后依次加入偶氮二異丁腈(AIBN)(4.93mg,0.03mmol)、PPEGMEMA-b-PDMAEMA(2790mg,0.3mmol)、甲基丙烯酸叔丁酯(tBMA)(1279.8mg,9mmol)、均三甲苯(100μl)及12ml甲苯,翻口橡皮塞密封。待反應(yīng)體系混合均勻后在冰水浴中用針頭通入氮氣除氧20min,將反應(yīng)瓶在65℃油浴鍋中反應(yīng)10h。反應(yīng)結(jié)束后將反應(yīng)液在正己烷中沉淀兩次,離心后真空干燥得到嵌段共聚物PPEGMEMA-b-PDMAEMA-b-PtBMA。通過核磁氫譜分析單體轉(zhuǎn)化率79.08%,理論分子量12673,所得三嵌段聚合物HLB值為14.7。
4)利用RAFT反應(yīng)合成具有核殼結(jié)構(gòu)的聚甲基丙烯酸叔丁酯納米球
將三嵌段聚合物PPEGMEMA-b-PDMAEMA-b-PtBMA(50mg)溶于10ml去離子水中作為水相,甲基丙烯酸叔丁酯(368mg)、乙二醇二甲基丙烯酸酯(92mg)、AIBN(0.32mg)和均三甲苯(10μl)作為油相。將油相和水相在25ml的玻璃瓶中混合均勻后,用超聲波破碎儀在60%振幅下超聲10min制成微乳液,所得微乳液轉(zhuǎn)入帶有橡皮塞的單口瓶,充氮氣除氧30min后升溫開始聚合,反應(yīng)溫度為75℃,反應(yīng)18h后得到具有核殼結(jié)構(gòu)的聚甲基丙烯酸叔丁酯納米球,單體轉(zhuǎn)化率100%,平均粒徑185nm。用無水乙醇和去離子水依次透析納米球溶液,真空凍干后得到聚甲基丙烯酸叔丁酯納米球凍干粉。
5)將聚甲基丙烯酸叔丁酯納米球凍干粉(100mg)加入90%(體積比)的三氟乙酸水溶液中,室溫下反應(yīng)過夜以脫除叔丁基,然后將反應(yīng)混合物去離子水中透析24h,將透析袋里的納米球溶液用真空凍干機干燥,得到具有擠壓和開關(guān)效應(yīng)的pH響應(yīng)型聚甲基丙烯酸納米球凍干粉。聚甲基丙烯酸叔丁酯納米球水解前后的透射電鏡圖見圖2,可以看出納米球具有明顯的核殼結(jié)構(gòu);水解前后的紅外光譜圖見圖3,可以看出水解后叔丁基在1392cm-1和1367cm-1處的特征峰消失,同時在3350cm-1處出現(xiàn)了一個很寬的羧基共軛峰;水解后納米粒子粒徑及zeta電勢隨pH變化圖見圖4,可以看出納米粒子具有明顯的pH響應(yīng)性。
6)載藥
將阿霉素(3mg)溶于5ml pH7.0的磷酸鹽緩沖液中,然后加入納米球凍干粉(5mg)在25℃水浴搖床中震蕩混合24h,離心分離除去上清液即得負(fù)載阿霉素的pH響應(yīng)型聚甲基丙烯酸納米球。載藥量為565.8μg/mg納米球,載藥率94.3%。
實施例2
1)利用RAFT反應(yīng)合成聚乙二醇單甲醚聚甲基丙烯酸酯(PPEGMEMA)
室溫下在50ml單口反應(yīng)瓶中加入攪拌子,然后依次加入偶氮二異丁腈(AIBN)(49.26mg,0.3mmol)、聚乙二醇單甲醚甲基丙烯酸酯(PEGMEMA)(9000mg,30mmol)、4-氰基-4-(苯基硫代甲酰硫基)戊酸(CPADB)(558.8mg,2mmol)、均三甲苯(100μl)及20ml甲苯,翻口橡皮塞密封。待反應(yīng)體系混合均勻后在冰水浴中用針頭通入氮氣除氧20min,將反應(yīng)瓶在65℃油浴鍋中反應(yīng)16h。反應(yīng)結(jié)束后將反應(yīng)液在正己烷中沉淀兩次,離心后真空干燥得到第一嵌段PPEGMEMA。通過核磁氫譜分析單體轉(zhuǎn)化率91.56%,理論分子量4400。
2)利用RAFT反應(yīng)合成聚乙二醇單甲醚聚甲基丙烯酸酯-聚甲基丙烯酸N,N-二甲胺基乙酯(PPEGMEMA-b-PDMAEMA)
室溫下在50ml單口反應(yīng)瓶中加入攪拌子,然后依次加入偶氮二異丁腈(AIBN)(4.11mg,0.025mmol)、PPEGMEMA(1100mg,0.25mmol)、甲基丙烯酸N,N-二甲氨基乙酯(DMAEMA)(3144mg,20mmol)、均三甲苯(100μl)及15ml甲苯,翻口橡皮塞密封。待反應(yīng)體系混合均勻后在冰水浴中用針頭通入氮氣除氧20min,將反應(yīng)瓶在65℃油浴鍋中反應(yīng)17h。反應(yīng)結(jié)束后將反應(yīng)液在正己烷中沉淀兩次,離心后真空干燥得到嵌段共聚物PPEGMEMA-PDMAEMA。通過核磁氫譜分析單體轉(zhuǎn)化率66.12%,理論分子量12715。
3)利用RAFT反應(yīng)合成聚乙二醇單甲醚聚甲基丙烯酸酯-聚甲基丙烯酸N,N-二甲胺基乙酯-聚甲基丙烯酸叔丁酯(PPEGMEMA-b-PDMAEMA-b-PtBMA)
室溫下在50ml單口反應(yīng)瓶中加入攪拌子,然后然后依次加入偶氮二異丁腈(AIBN)(3.28mg,0.02mmol)、PPEGMEMA-b-PDMAEMA(2543mg,0.2mmol)、甲基丙烯酸叔丁酯(tBMA)(1137.6mg,8mmol)、均三甲苯(100μl)及12ml甲苯,翻口橡皮塞密封。待反應(yīng)體系混合均勻后在冰水浴中用針頭通入氮氣除氧20min,將反應(yīng)瓶在65℃油浴鍋中反應(yīng)12h。反應(yīng)結(jié)束后將反應(yīng)液在正己烷中沉淀兩次,離心后真空干燥得到嵌段共聚物PPEGMEMA-b-PDMAEMA-b-PtBMA。通過核磁氫譜分析單體轉(zhuǎn)化率75.36%,理論分子量17001,所得三嵌段聚合物HLB值為14.95。
4)利用RAFT反應(yīng)合成具有核殼結(jié)構(gòu)的聚甲基丙烯酸叔丁酯納米球
將三嵌段聚合物PPEGMEMA-b-PDMAEMA-b-PtBMA(67mg)溶于10ml去離子水中作為水相,甲基丙烯酸叔丁酯(414mg)、乙二醇二甲基丙烯酸酯(46mg)、AIBN(0.32mg)和均三甲苯(10μl)作為油相。將油相和水相在25ml的玻璃瓶中混合均勻后,用超聲波破碎儀在65%振幅下超聲10min制成微乳液,所得微乳液轉(zhuǎn)入帶有橡皮塞的單口瓶,充氮氣除氧30min后升溫開始聚合,反應(yīng)溫度為75℃,反應(yīng)18h后得到具有核殼結(jié)構(gòu)的聚甲基丙烯酸叔丁酯納米球,單體轉(zhuǎn)化率100%,平均粒徑205nm。用無水乙醇和去離子水依次透析納米球溶液,真空凍干后得到聚甲基丙烯酸叔丁酯納米球凍干粉。
5)將聚甲基丙烯酸叔丁酯納米球凍干粉(100mg)加入90%(體積比)的三氟乙酸水溶液中,室溫下反應(yīng)過夜以脫除叔丁基,然后將反應(yīng)混合物去離子水中透析24h,將透析袋里的納米球溶液用真空凍干機干燥,得到具有擠壓和開關(guān)效應(yīng)的pH響應(yīng)型聚甲基丙烯酸納米球凍干粉。
6)載藥
將阿霉素(5.0mg)溶于5ml pH7.0的磷酸鹽緩沖液中,然后加入納米球凍干粉(5mg)在25℃水浴搖床中震蕩混合24h,離心分離除去上清液即得負(fù)載阿霉素的pH響應(yīng)型聚甲基丙烯酸納米球。載藥量為754.6μg/mg納米球,載藥率75.5%。
實施例3
1)利用RAFT反應(yīng)合成聚乙二醇單甲醚聚丙烯酸酯(PPEGMEA)
室溫下在25ml單口反應(yīng)瓶中加入攪拌子,然后依次加入偶氮二異丁腈(AIBN)(2.46mg,0.015mmol)、聚乙二醇單甲醚丙烯酸酯(PEGMEA)(2880mg,6mmol)、4-氰基-4-(丙基三硫代碳酸酯基)戊酸(CPPT)(41.6mg,0.15mmol)、均三甲苯(100μl)及5ml甲苯,翻口橡皮塞密封。待反應(yīng)體系混合均勻后在冰水浴中用針頭通入氮氣除氧20min,將反應(yīng)瓶在65℃油浴鍋中反應(yīng)16h。反應(yīng)結(jié)束后將反應(yīng)液在正己烷中沉淀兩次,離心后真空干燥得到第一嵌段PPEGMEA。通過核磁氫譜分析單體轉(zhuǎn)化率93.61%,理論分子量18250。
2)利用RAFT反應(yīng)合成聚乙二醇單甲醚聚丙烯酸酯-聚丙烯酸N,N-二乙胺基乙酯(PPEGMEA-b-PDEAEA)
室溫下在50ml單口反應(yīng)瓶中加入攪拌子,然后依次加入偶氮二異丁腈(AIBN)(4.11mg,0.025mmol)、PPEGMEMA(4563mg,0.25mmol)、丙烯酸N,N-二乙氨基乙酯(DEAEA)(3424mg,20mmol)、均三甲苯(100μl)及15ml甲苯,翻口橡皮塞密封。待反應(yīng)體系混合均勻后在冰水浴中用針頭通入氮氣除氧20min,將反應(yīng)瓶在65℃油浴鍋中反應(yīng)10h。反應(yīng)結(jié)束后將反應(yīng)液在正己烷中沉淀兩次,離心后真空干燥得到嵌段共聚物PPEGMEA-PDEAEA。通過核磁氫譜分析單體轉(zhuǎn)化率57.31%,理論分子量26100。
3)利用RAFT反應(yīng)合成聚乙二醇單甲醚丙烯酸酯-聚丙烯酸N,N-二乙胺基乙酯-聚丙烯酸叔丁酯(PPEGMEA-b-PDEAEA-b-PtBA)
室溫下在50ml單口反應(yīng)瓶中加入攪拌子,然后然后依次加入偶氮二異丁腈(AIBN)(3.28mg,0.02mmol)、PPEGMEA-b-PDEAEA(5220mg,0.2mmol)、丙烯酸叔丁酯(tBA)(2563.4mg,20mmol)、均三甲苯(100μl)及15ml甲苯,翻口橡皮塞密封。待反應(yīng)體系混合均勻后在冰水浴中用針頭通入氮氣除氧20min,將反應(yīng)瓶在65℃油浴鍋中反應(yīng)19h。反應(yīng)結(jié)束后將反應(yīng)液在正己烷中沉淀兩次,離心后真空干燥得到嵌段共聚物PPEGMEA-b-PDMAEA-b-PtBA。通過核磁氫譜分析單體轉(zhuǎn)化率87.27%,理論分子量37199,所得三嵌段聚合物HLB值為14.03。
4)利用RAFT反應(yīng)合成具有核殼結(jié)構(gòu)的聚丙烯酸叔丁酯納米球
將三嵌段聚合物PPEGMEA-b-PDEAEA-b-PtBA(106mg)溶于10ml去離子水中作為水相,丙烯酸叔丁酯(322mg)、乙二醇二丙烯酸酯(138mg)、AIBN(0.47mg)和均三甲苯(10μl)作為油相。將油相和水相在25ml的玻璃瓶中混合均勻后,用超聲波破碎儀在70%振幅下超聲10min制成微乳液,所得微乳液轉(zhuǎn)入帶有橡皮塞的單口瓶,充氮氣除氧30min后升溫開始聚合,反應(yīng)溫度為75℃,反應(yīng)18h后得到具有核殼結(jié)構(gòu)的聚丙烯酸叔丁酯納米球,單體轉(zhuǎn)化率100%,平均粒徑223nm。用無水乙醇和去離子水依次透析納米球溶液,真空凍干后得到聚丙烯酸叔丁酯納米球凍干粉。
5)將聚丙烯酸叔丁酯納米球凍干粉(100mg)加入90%(體積比)的三氟乙酸水溶液中,室溫下反應(yīng)過夜以脫除叔丁基,然后將反應(yīng)混合物去離子水中透析24h,將透析袋里的納米球溶液用真空凍干機干燥,得到具有擠壓和開關(guān)效應(yīng)的pH響應(yīng)型聚丙烯酸納米球凍干粉。
6)載藥
將表柔比星(1.5mg)溶于5ml pH7.0的磷酸鹽緩沖液中,然后加入納米球凍干粉(5mg)在25℃水浴搖床中震蕩混合24h,離心分離除去上清液即得負(fù)載表柔比星的pH響應(yīng)型聚丙烯酸納米球。載藥量為236.4μg/mg納米球,載藥率為78.8%。
實施例4
1)利用RAFT反應(yīng)合成聚乙二醇單甲醚聚甲基丙烯酸酯(PPEGMEMA)
室溫下在50ml單口反應(yīng)瓶中加入攪拌子,然后依次加入偶氮二異丁腈(AIBN)(16.42mg,0.1mmol)、聚乙二醇單甲醚甲基丙烯酸酯(PEGMEMA)(9000mg,30mmol)、4-氰基-4-(丙基三硫代碳酸酯基)戊酸(CPPT)(277mg,1mmol)、均三甲苯(100μl)及30ml甲苯,翻口橡皮塞密封。待反應(yīng)體系混合均勻后在冰水浴中用針頭通入氮氣除氧20min,將反應(yīng)瓶在60℃油浴鍋中反應(yīng)15h。反應(yīng)結(jié)束后將反應(yīng)液在正己烷中沉淀兩次,離心后真空干燥得到第一嵌段PPEGMEMA。通過核磁氫譜分析單體轉(zhuǎn)化率90.45%,理論分子量8418。
2)利用RAFT反應(yīng)合成聚乙二醇單甲醚聚甲基丙烯酸酯-聚甲基丙烯酸N,N-二甲胺基乙酯(PPEGMEMA-b-PDMAEMA)
室溫下在25ml單口反應(yīng)瓶中加入攪拌子,然后依次加入偶氮二異丁腈(AIBN)(4.93mg,0.03mmol)、PPEGMEMA(2525.4mg,0.3mmol)、甲基丙烯酸N,N-二甲氨基乙酯(DMAEMA)(2830mg,18mmol)、均三甲苯(100μl)及12ml甲苯,翻口橡皮塞密封。待反應(yīng)體系混合均勻后在冰水浴中用針頭通入氮氣除氧20min,將反應(yīng)瓶在70℃油浴鍋中反應(yīng)13h。反應(yīng)結(jié)束后將反應(yīng)液在正己烷中沉淀兩次,離心后真空干燥得到嵌段共聚物PPEGMEMA-PDMAEMA。通過核磁氫譜分析單體轉(zhuǎn)化率61.52%,理論分子量14221。
3)利用RAFT反應(yīng)合成聚乙二醇單甲醚聚甲基丙烯酸酯-聚甲基丙烯酸N,N-二甲胺基乙酯-聚甲基丙烯酸叔丁酯(PPEGMEMA-b-PDMAEMA-b-PtBMA)
室溫下在50ml單口反應(yīng)瓶中加入攪拌子,然后然后依次加入偶氮二異丁腈(AIBN)(4.93mg,0.03mmol)、PPEGMEMA-b-PDMAEMA(4266.3mg,0.3mmol)、甲基丙烯酸叔丁酯(tBMA)(3412.8mg,24mmol)、均三甲苯(150μl)及24ml甲苯,翻口橡皮塞密封。待反應(yīng)體系混合均勻后在冰水浴中用針頭通入氮氣除氧20min,將反應(yīng)瓶在70℃油浴鍋中反應(yīng)12h。反應(yīng)結(jié)束后將反應(yīng)液在正己烷中沉淀兩次,離心后真空干燥得到嵌段共聚物PPEGMEMA-b-PDMAEMA-b-PtBMA。通過核磁氫譜分析單體轉(zhuǎn)化率75.01%,理論分子量22754,所得三嵌段聚合物HLB值為12.5。
4)利用RAFT反應(yīng)合成具有核殼結(jié)構(gòu)的聚甲基丙烯酸叔丁酯納米球
將三嵌段聚合物PPEGMEMA-b-PDMAEMA-b-PtBMA(90mg)溶于10ml去離子水中作為水相,甲基丙烯酸叔丁酯(391mg)、乙二醇二甲基丙烯酸酯(69mg)、AIBN(0.33mg)和均三甲苯(10μl)作為油相。將油相和水相在25ml的玻璃瓶中混合均勻后,用超聲波破碎儀在65%振幅下超聲15min制成微乳液,所得微乳液轉(zhuǎn)入帶有橡皮塞的單口瓶,充氮氣除氧30min后升溫開始聚合,反應(yīng)溫度為80℃,反應(yīng)15h后得到具有核殼結(jié)構(gòu)的聚甲基丙烯酸叔丁酯納米球,單體轉(zhuǎn)化率100%,平均粒徑187nm。用無水乙醇和去離子水依次透析納米球溶液,真空凍干后得到聚甲基丙烯酸叔丁酯納米球凍干粉。
5)將聚甲基丙烯酸叔丁酯納米球凍干粉(100mg)加入90%(體積比)的三氟乙酸水溶液中,室溫下反應(yīng)過夜以脫除叔丁基,然后將反應(yīng)混合物去離子水中透析24h,將透析袋里的納米球溶液用真空凍干機干燥,得到具有擠壓和開關(guān)效應(yīng)的pH響應(yīng)型聚甲基丙烯酸納米球凍干粉。
6)載藥
將吉西他濱(2mg)溶于5ml pH7.0的磷酸鹽緩沖液中,然后加入納米球凍干粉(5mg)在25℃水浴搖床中震蕩混合24h,離心分離除去上清液即得負(fù)載阿霉素的pH響應(yīng)型聚甲基丙烯酸納米球。載藥量為352.9μg/mg納米球,載藥率為88.2%。
實施例5
1)利用RAFT反應(yīng)合成聚乙二醇單甲醚聚丙烯酸酯(PPEGMEA)
室溫下在25ml單口反應(yīng)瓶中加入攪拌子,然后依次加入偶氮二異丁腈(AIBN)(3.28mg,0.02mmol)、聚乙二醇單甲醚丙烯酸酯(PEGMEA)(1920mg,4mmol)、2-氰基-2-丙基苯并二硫(CPDB)(44.3mg,0.2mmol)、均三甲苯(100μl)及5ml甲苯,翻口橡皮塞密封。待反應(yīng)體系混合均勻后在冰水浴中用針頭通入氮氣除氧20min,將反應(yīng)瓶在70℃油浴鍋中反應(yīng)16h。反應(yīng)結(jié)束后將反應(yīng)液在正己烷中沉淀兩次,離心后真空干燥得到第一嵌段PPEGMEA。通過核磁氫譜分析單體轉(zhuǎn)化率95.82%,理論分子量9420。
2)利用RAFT反應(yīng)合成聚乙二醇單甲醚聚丙烯酸酯-聚丙烯酸N,N-二甲胺基乙酯(PPEGMEA-b-PDMAEA)
室溫下在25ml單口反應(yīng)瓶中加入攪拌子,然后依次加入偶氮二異丁腈(AIBN)(4.11mg,0.025mmol)、PPEGMEMA(2355mg,0.25mmol)、丙烯酸N,N-二甲氨基乙酯(DMAEA)(1790mg,12.5mmol)、均三甲苯(100μl)及6.25ml甲苯,翻口橡皮塞密封。待反應(yīng)體系混合均勻后在冰水浴中用針頭通入氮氣除氧20min,將反應(yīng)瓶在70℃油浴鍋中反應(yīng)19h。反應(yīng)結(jié)束后將反應(yīng)液在正己烷中沉淀兩次,離心后真空干燥得到嵌段共聚物PPEGMEA-PDMAEA。通過核磁氫譜分析單體轉(zhuǎn)化率75.13%,理論分子量14800。
3)利用RAFT反應(yīng)合成聚乙二醇單甲醚丙烯酸酯-聚丙烯酸N,N-二甲胺基乙酯-聚丙烯酸叔丁酯(PPEGMEA-b-PDMAEA-b-PtBA)
室溫下在50ml單口反應(yīng)瓶中加入攪拌子,然后然后依次加入偶氮二異丁腈(AIBN)(3.28mg,0.02mmol)、PPEGMEA-b-PDMAEA(2960mg,0.2mmol)、丙烯酸叔丁酯(tBA)(1923mg,15mmol)、均三甲苯(100μl)及15ml甲苯,翻口橡皮塞密封。待反應(yīng)體系混合均勻后在冰水浴中用針頭通入氮氣除氧20min,將反應(yīng)瓶在60℃油浴鍋中反應(yīng)12h。反應(yīng)結(jié)束后將反應(yīng)液在正己烷中沉淀兩次,離心后真空干燥得到嵌段共聚物PPEGMEA-b-PDMAEA-b-PtBA。通過核磁氫譜分析單體轉(zhuǎn)化率57.64%,理論分子量20342,所得三嵌段聚合物HLB值為14.55。
4)利用RAFT反應(yīng)合成具有核殼結(jié)構(gòu)的聚丙烯酸叔丁酯納米球
將三嵌段聚合物PPEGMEA-b-PDMAEA-b-PtBA(86mg)溶于10ml去離子水中作為水相,丙烯酸叔丁酯(345mg)、乙二醇二丙烯酸酯(115mg)、AIBN(0.69mg)和均三甲苯(10μl)作為油相。將油相和水相在25ml的玻璃瓶中混合均勻后,用超聲波破碎儀在70%振幅下超聲15min制成微乳液,所得微乳液轉(zhuǎn)入帶有橡皮塞的單口瓶,充氮氣除氧30min后升溫開始聚合,反應(yīng)溫度為75℃,反應(yīng)18h后得到具有核殼結(jié)構(gòu)的聚丙烯酸叔丁酯納米球,單體轉(zhuǎn)化率100%,平均粒徑173nm。用無水乙醇和去離子水依次透析納米球溶液,真空凍干后得到聚丙烯酸叔丁酯納米球凍干粉。
5)將聚丙烯酸叔丁酯納米球凍干粉(100mg)加入90%(體積比)的三氟乙酸水溶液中,室溫下反應(yīng)過夜以脫除叔丁基,然后將反應(yīng)混合物去離子水中透析24h,將透析袋里的納米球溶液用真空凍干機干燥,得到具有擠壓和開關(guān)效應(yīng)的pH響應(yīng)型聚丙烯酸納米球凍干粉。
6)載藥
將阿霉素(3mg)溶于5ml pH7.0的磷酸鹽緩沖液中,然后加入納米球凍干粉(5mg)在25℃水浴搖床中震蕩混合24h,離心分離除去上清液即得負(fù)載表柔比星的pH響應(yīng)型聚丙烯酸納米球。載藥量為493.9μg/mg納米球,載藥率為82.3%。
實驗例
為了驗證本發(fā)明中載藥納米球的擠壓和開關(guān)功能,我們查了實施例1中所制備的載藥納米球的藥物釋放曲線,結(jié)果如圖5所示。圖中a線為阿霉素在pH5.0時的藥物釋放曲線,b線為阿霉素在pH7.4時的藥物釋放曲線。在核層的迅速收縮擠壓(pH5.0)和殼層的收縮封閉(pH7.4)協(xié)同作用下,阿霉素在pH5.0時的藥物釋放速率是在pH7.4時的3倍左右。
- 還沒有人留言評論。精彩留言會獲得點贊!
- 一種基于納米效應(yīng)的巖土材料性能提高方法及裝置的制造方法
- 一種具有電氧化催化性能的磁性納米多孔Fe-Pt合金的制備方法
- 一種FeP/石墨烯復(fù)合材料及其制備方法
- 一種多巴胺類化合物修飾或包裹納米粒子改性聚合物復(fù)合材料及其制備方法
- 一種納米銀/石墨烯復(fù)合材料及其制備方法
- 適用于納米尺寸下工藝不敏感的高增益兩級運算放大器的制造方法
- 一種尺寸可控納米級立方相超細(xì)鈦酸鋇粉體的制備方法
- 一種低溫合成高純納米錳酸鋰的工藝方法
- 一種具有花瓣效應(yīng)的ZnO納米結(jié)構(gòu)的制備方法
- 一種利用咖啡環(huán)效應(yīng)用于制備致密納米顆粒薄膜和有序納米線薄膜的方法
- 一種納米固體反應(yīng)器的制備方法
- 納米圖形化中凸起效應(yīng)的去除的制作方法
- 一種具有納米效應(yīng)催化子網(wǎng)群結(jié)構(gòu)的水面凈化裝置的制作方法
- 一種具有納米效應(yīng)催化元結(jié)構(gòu)的飲品包裝體的制作方法
- 多質(zhì)離散效應(yīng)納米結(jié)構(gòu)液膜及其制備方法和應(yīng)用的制作方法
- 能夠補償納米形貌效應(yīng)的化學(xué)機械拋光用漿液組合物、以及利用其的半導(dǎo)體元件的表面 ...的制作方法
- 一種納米改性雙透效應(yīng)彈性體材料制作方法
- 一種基于電致發(fā)熱效應(yīng)的一維納米材料的焊接方法
- 由電效應(yīng)控制電活性材料厚度變化實現(xiàn)局部變形的反射鏡的制作方法
- 具有ct示蹤效應(yīng)的可生物降解納米藥物膠囊及其制備方法
- 一種基于微通道與細(xì)胞表面觸碰效應(yīng)的細(xì)胞分析裝置及方法
- 在特種產(chǎn)品基材表面制備微納米級別金屬電極的方法
- 一種強永磁性納米多孔Fe-Pt合金及其制備方法
- 一種鋼件表面納米化低溫滲鋁處理方法
- 一種竹材表面水熱法生長納米ZnO互穿網(wǎng)絡(luò)結(jié)構(gòu)的制備方法
- 一種在納米赤鐵礦表面形成的Fe-O活性物種及其制備方法和應(yīng)用
- 一種用于航天器表面帶電效應(yīng)的監(jiān)測裝置的制造方法
- 具有親水表面的由納米片組成的硒化鎘花狀微球及其制備方法和應(yīng)用
- 在多晶硅上形成倒金字塔型多孔表面納米織構(gòu)的方法及制備短波增強型太陽電池的方法
- 一種在發(fā)光二極管表面制備納米尺度球形體結(jié)構(gòu)的方法
- 具跨尺度多相原位增強效應(yīng)的耐熱合金鋼成分及其微結(jié)構(gòu)調(diào)控工藝的制作方法
- 一種帶有納米尺度通道的su-8膠電液動力射流噴針制造方法
- 納米尺度模板結(jié)構(gòu)上的ⅲ族-n晶體管的制作方法
- 一種碳納米管/聚酯復(fù)合材料制備方法
- 納米尺度的微細(xì)圖形加工方法
- 具有納米尺度凸起結(jié)構(gòu)的無紡布及其制造方法和應(yīng)用
- 靜電紡絲制備纖維素基納米復(fù)合纖維薄膜的方法
- 一種適于納米尺度工藝的高線性度射頻前端的制作方法
- 一種納米尺度鋁基金屬-有機框架結(jié)構(gòu)材料及其制備方法
- 基于納米尺度線的數(shù)據(jù)存儲的制作方法
- 鰭式場效應(yīng)晶體管及其形成方法與制造工藝
- 一種具有擠壓和開關(guān)效應(yīng)的納米藥物載體的制備方法與制造工藝
- 一種穩(wěn)定的以氣體為絕緣層的納米線場效應(yīng)晶體管的制作方法
- 鰭式碳納米管場效應(yīng)晶體管的制作方法
- 一種納米柱結(jié)構(gòu)有機場效應(yīng)晶體管存儲器及其制備方法
- 一種納米晶體氣動儀測量頭的制作方法
- 利用碳納米場效應(yīng)晶體管實現(xiàn)的三值靜態(tài)隨機存儲單元的制作方法
- 一種超強超高熱穩(wěn)定性塊體納米晶鋼的制備方法
- 一種以碳量子點為晶種制備Fe納米顆粒的方法
- 納米線場效應(yīng)晶體管(fet)器件及其制造方法