一種DAP-seq測序建庫方法與流程
本發(fā)明涉及高通量測序建庫,特別涉及一種dap-seq測序建庫方法。
背景技術(shù):
1、轉(zhuǎn)錄因子(transcription?factor,tf)在基因表達調(diào)控中扮演著核心角色,通過特異性結(jié)合到基因組上的轉(zhuǎn)錄因子結(jié)合位點(transcriptionfactor?binding?sites,tfbs)來調(diào)控下游基因的表達。探究tfbs對于理解tf的功能具有重要意義。染色質(zhì)免疫沉淀測序(chip-seq)是一種揭示tfbs的有效手段。然而,chip-seq的成功在很大程度上依賴于抗體的質(zhì)量。對于稀有或表達水平較低的蛋白質(zhì),制備高質(zhì)量的抗體具有一定難度。此外,對于非模式生物,可用的商業(yè)化抗體資源極其有限。這些因素限制了chip-seq技術(shù)在非模式生物或不太常見的轉(zhuǎn)錄因子中的廣泛應(yīng)用。
2、dna親和純化測序(dnaaffinitypurification?sequencing,dap-seq)克服了chip-seq在抗體獲取和物種方面的限制,允許研究者在體外條件下,利用經(jīng)過特殊改造的轉(zhuǎn)錄因子蛋白直接與基因組dna片段相互作用,對富集的dna片段進行高通量測序,以識別tfbs。dap-seq技術(shù)因其不依賴抗體和物種的優(yōu)勢,自推出后,已被廣泛應(yīng)用于多個學(xué)科,有效助力了轉(zhuǎn)錄調(diào)控機制的深入解析。
3、盡管dap-seq技術(shù)在tfbs的研究中提供了一種有效的策略,但在實際的質(zhì)粒構(gòu)建階段,研究人員仍面臨著難題。在質(zhì)粒的構(gòu)建初期,有時需要確定合適的限制性內(nèi)切酶對載體和外源dna進行切割,設(shè)計合適的改造轉(zhuǎn)錄因子蛋白和相應(yīng)的dna結(jié)合序列可能較為復(fù)雜。在質(zhì)粒構(gòu)建過程中,效率也不如預(yù)期,需要多次的構(gòu)建才能成功,并且質(zhì)粒的提取、純化、驗證過程往往耗時較長,甚至有假陽性克隆的可能,這些情況無形中增加了研究者實驗與時間的成本。
技術(shù)實現(xiàn)思路
1、本發(fā)明提供了一種dap-seq測序建庫方法,對傳統(tǒng)的dna親和純化測序(dap-seq)實驗流程進行優(yōu)化,提高了質(zhì)粒構(gòu)建的效率,減少所需的時間和資源消耗。
2、本發(fā)明提供了一種dap-seq測序建庫方法,包括:
3、s1、設(shè)計基因特異性引物,以使所述引物能夠精準(zhǔn)識別并結(jié)合到目標(biāo)基因序列上,通過pcr擴增基因片段并進行純化,以確保獲得高純度的目標(biāo)dna作為體外蛋白翻譯的基因模板;
4、s2、在體外蛋白表達階段,利用特定的表達系統(tǒng),投入模板合成目標(biāo)蛋白質(zhì);
5、s3、所述蛋白質(zhì)在dna片段的篩選過程中,與特定dna結(jié)合,利用蛋白質(zhì)與dna之間的相互作用,通過親和純化技術(shù)篩選出dna片段;
6、s4、dna片段被構(gòu)建成文庫上機測序。
7、進一步地,所述步驟s1具體包括:
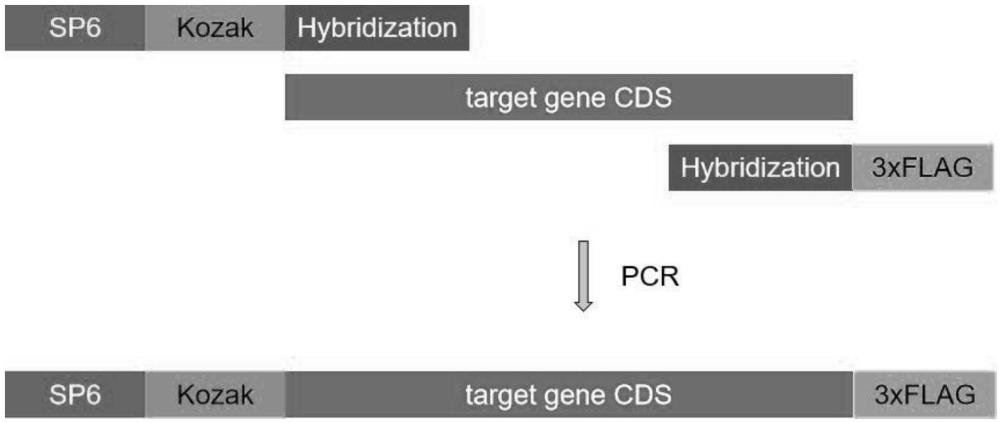
8、s11、將3xflag標(biāo)簽連接于目標(biāo)蛋白c端或n端,其中:
9、c端:上游引物f包括保護堿基、sp6啟動子、轉(zhuǎn)錄起始位點、kozak序列、翻譯起始密碼子、目的蛋白cds區(qū)起始位置17-22bp序列;下游引物r包括翻譯終止密碼子反向互補序列、3xflag反向互補序列、目的蛋白cds終止密碼子之前序列的反向互補序列;
10、n端:上游引物f包括保護堿基、sp6啟動子、轉(zhuǎn)錄起始位點、kozak序列、翻譯起始密碼子、3xflag序列、目的蛋白cds區(qū)起始密碼子atg之后的序列;下游引物r包括翻譯終止密碼子反向互補序列和目的蛋白cds區(qū)終止密碼子之前序列的反向互補序列;
11、s12、引物測試與比對:利用pcr擴增目的片段后,使用一代sanger測序獲得序列信息并比對確認;
12、s13、擴增與純化:根據(jù)引物擴增得到pcr產(chǎn)物,進行瓊脂糖凝膠電泳,利用膠回收試劑盒回收純化pcr產(chǎn)物。
13、進一步地,所述步驟s2具體包括:
14、將解凍后的30μl體外表達蛋白試劑盒中的master?mix與5~8μl?pcr產(chǎn)物配置反應(yīng)混合液,并用ddh2o將體系補至50μl,吸打混勻后于25℃孵育2h。
15、進一步地,所述步驟s3中,dna片段化及片段篩選具體包括:
16、s301、取100pg~1μg樣品dna,加入5μl?feabuffer,并用ddh2o補至40μl,全程于冰上操作;接著加入10μl?fea?enzyme?mix,使用移液器吹打或振蕩混勻,并短暫離心將反應(yīng)液收集至管底,置于pcr儀中進行反應(yīng);
17、s302、根據(jù)dna量按稀釋adapter至合適濃度,按照第一預(yù)設(shè)表配置連接反應(yīng)體系;
18、s303、使用移液器吹打混勻,并短暫離心后將pcr管置于pcr儀中;
19、s304、將平衡至室溫的80μl?dnaclean?beads加入上述pcr反應(yīng)產(chǎn)物,吸打混勻后于室溫下靜置10min;
20、s305、將pcr管開蓋、置于磁力架上,靜置5min,棄上清;
21、s306、保持pcr管始終置于磁力架上,加200μl新鮮配置80%乙醇,室溫孵育30s后,移除上清,重復(fù)一次后,開蓋空氣干燥磁珠5min;
22、s307、將pcr管從磁力架中取出,加入23μl?h2o,重懸磁珠,室溫靜置2min;
23、s308、將pcr管開蓋置于磁力架上,靜置5min后,小心吸取21μl上清至一個新的pcr管中,加入2μl的dap?f和2μl的dap?r,至總體積25μl,置于pcr儀中,擴增文庫:
24、s309、在產(chǎn)物中加入40μl(0.8x)dnaclean?beads,吸打混勻后室溫放置10min;
25、s310、重復(fù)步驟s305-s307;
26、s311、將pcr管開蓋置于磁力架上,靜置5min后,小心吸取21μl上清至一個新的pcr管中。
27、進一步地,所述第一預(yù)設(shè)表中包括組分和每個組分的體積,具體為:上一步產(chǎn)物50μl、rapid?ligation?buffer?325μl、rapid?dnaligase?5μl、dapadapter?5μl、ddh2o?15μl、total?100μl。
28、進一步地,所述步驟s3中,蛋白親和吸附dna片段具體包括:
29、s3001、轉(zhuǎn)移20μl已平衡至室溫的抗體磁珠至ep管,置磁力架,貼壁1min,溶液澄清后,棄上清;
30、s3002、將ep管從磁力架上取出,加入20μl?dapbuffer,重懸磁珠,洗滌干凈后,置磁力架,貼壁1min,溶液澄清后,棄上清;
31、s3003、加入40μl?dap?buffer,重懸磁珠,加40μl蛋白體外表達混合液,重懸磁珠,置于旋轉(zhuǎn)混合儀上,25℃孵育1h;
32、s3004、將剩余10μl轉(zhuǎn)移至新ep管,標(biāo)記為input-protein;
33、s3005、完成孵育后,閃甩,使液體和磁珠均位于管底,置于磁力架上,貼壁1min,上清標(biāo)記為supernatant,保存于-20℃;
34、s3006、加入85μl?dapbuffer重懸磁珠,磁珠自然沉降后,置于磁力架上,貼壁1min,棄上清;
35、s3007、加入40μl?dapbuffer重懸磁珠,加入30~100ng?input?dna,補ddh2o至80μl,置于旋轉(zhuǎn)混合儀上,4℃孵育過夜;
36、s3008、將ep管從旋轉(zhuǎn)混合儀取出,閃甩;置于磁力架上,貼壁1min,棄上清;
37、s3009、將ep管從磁力架上取出,加入85μl冰預(yù)冷的dapbuffer,重懸磁珠,洗滌干凈后轉(zhuǎn)移至1個新的ep管,加入17μl磁珠標(biāo)記為dap-protein,剩余磁珠標(biāo)記為dap-dna;
38、s3010、向s3004中input-protein管加入20μl?2x?sds?loading?buffer,從s3005中supernatant中分離20μl至新ep管,加入20μl?2x?sds?loading?buffer,將s3009中dap-protein管置于磁力架,靜置5min棄上清,加入25μl?1x?sds?loadingbuffer;
39、s3011、將上述3管煮沸10min后置磁力架,靜置5min后,將上清轉(zhuǎn)移到3個新的ep管中,進行western?blot質(zhì)檢;
40、s3012、將dap-dna管置于磁力架上,靜置5min,棄上清后加入200μl?elutionbuffer,重懸磁珠;input-dna樣品用elutionbuffer補足到200μl;
41、s3013、向上述每管中分別加入5μg?rnasea,thermo?mixer上37℃震蕩30min;
42、s3014、閃甩,每管分別加入200μg?proteinase?k,纏上封口膜,65℃震蕩2h;
43、s3015、將ep管置磁力架,靜置5min,將上清轉(zhuǎn)移到新的ep管中;
44、s3016、每管中加入200μl酚氯仿異戊醇(ph8.0)劇烈震蕩30s,室溫、12000rpm離心15min,轉(zhuǎn)移200μl上清到新的ep管中;
45、s3017、每管中加入20μl?3m?naac(ph5.2),1μl?glycogen,混勻,再加入600μl無水乙醇,劇烈倒置混勻,于-80℃沉淀1h;
46、s3018、沉淀結(jié)束后,4℃、12000rpm離心15min,棄掉乙醇;
47、s3019、加入1ml冰預(yù)冷的75%乙醇,緩慢倒置數(shù)次洗滌,4℃、12000rpm離心5min,棄乙醇;重復(fù)一次;
48、s3020、閃甩,徹底吸凈剩余乙醇,56℃干燥3min后,input加入50μl?h2o回溶,dap加入30μl?h2o回溶。
49、進一步地,所述步驟s4具體包括:
50、s41、按照第二預(yù)設(shè)表配制擴增反應(yīng)混合液,加入到0.2mlpcr管中;
51、s42、將樣品置于pcr儀中,擴增文庫;
52、s43、將pcr產(chǎn)物補水至100μl,混勻后加入70μl(0.7x)dnaclean?beads,吹打混勻,室溫放置10min;
53、s44、將pcr管開蓋、置于磁力架上,靜置5min后轉(zhuǎn)移上清至新的pcr管中,加入20μl(0.2x)dnaclean?beads,吹打混勻,室溫放置10min,開蓋再靜置5min;
54、s45、保持pcr管始終置于磁力架上,加入200μl新鮮配置80%乙醇,室溫孵育30s后,移除上清;該步驟重復(fù)操作一次;開蓋空氣干燥磁珠5min;
55、s46、將pcr管從磁力架中取出,加入27μl?h2o,重懸磁珠,室溫靜置2min;
56、s47、將pcr管開蓋置于磁力架上,靜置5min后,吸取25μl上清至一個新的pcr管中;
57、s48使用qubit試劑對文庫進行定量,用bioanalyzer進行片段分析。
58、進一步地,所述第二預(yù)設(shè)表中包括組分和每個組分的體積,具體為:input?or?dapdnaxμl、2x?kapahifi?hotstart?readymix?25μl、bgi?f引物(10μm)2.5μl、bgi?rx引物(10μm)2.5μl、ddh2o補至總體積50μl。
59、本發(fā)明的有益效果為:
60、1、減少克隆過程中的錯誤:傳統(tǒng)的質(zhì)粒構(gòu)建方法涉及到多個步驟,如限制性酶切、連接、轉(zhuǎn)化等,每個步驟都可能引入錯誤或變異。而新方法通過直接表達pcr產(chǎn)物,減少了這些步驟,從而降低了因克隆過程中錯誤而導(dǎo)致的實驗誤差。
61、2、提高實驗效率:由于省去了質(zhì)粒構(gòu)建和驗證的時間,本發(fā)明可以更快地獲得實驗數(shù)據(jù),這對于需要快速得到結(jié)果的研究尤為重要。
62、3、減少假陽性結(jié)果:傳統(tǒng)方法中,質(zhì)粒構(gòu)建的不穩(wěn)定性可能導(dǎo)致假陽性克隆的出現(xiàn)。本發(fā)明通過直接表達,減少了這種可能性,提高了結(jié)果的可靠性。
63、4、簡化操作流程:簡化的實驗流程減少了人為操作錯誤的可能性,從而提高了實驗的重復(fù)性和一致性。
64、5、成本效益:由于省去了質(zhì)粒構(gòu)建和驗證的成本,本發(fā)明在經(jīng)濟上更為高效,使得更多的研究者能夠進行dap-seq實驗。
65、6、靈活性:本發(fā)明允許快速更換不同的dna片段進行測試,這為研究者提供了更大的靈活性,可以更廣泛地探索不同的tf和dna結(jié)合位點。
- 還沒有人留言評論。精彩留言會獲得點贊!