一種9?去甲基?α?二氫丁苯那嗪及其雜質的分離測定方法與流程
本發明屬于藥物分析領域,具體涉及一種9-去甲基-α-二氫丁苯那嗪及其雜質的分離測定方法。
背景技術:
Ⅱ型囊泡單胺轉運體(VMAT2)是一種位于神經元胞質內囊泡膜上的膜蛋白,在單胺類遞質的重攝取過程中起著關鍵作用。分子克隆技術鑒定表明,VMAT2主要分布于人類及哺乳動物的中樞神經系統與胰臟組織內。VMAT2的變化與多種疾病有關,如帕金森病(PD)、阿爾茨海默病(AD)、亨廷頓病(HD)、糖尿病等。近年來,以VMAT2為靶標進行放射性示蹤顯像,對相關疾病進行早期診斷、分級、鑒別診斷、療效監測研究,成為核醫學研究的熱點之一。到目前為止,國內外已成功應用于臨床的VMAT2顯像劑,都是用于正電子發射計算機斷層(PET)成像的正電子示蹤藥物,如:11C-α-DTBZ、18F-FP-α-DTBZ等。
9-去甲基-α-二氫丁苯那嗪(9-DM-α-DTBZ)是制備PET顯像藥物11C-α-DTBZ的標記前體化合物,標記過程如下所示:
由于11C半衰期僅為20min,臨床上使用示蹤藥物11C-α-DTBZ需要通過標記前體化合物9-DM-α-DTBZ現場制備而得。標記前體化合物9-DM-α- DTBZ中雜質的含量影響到PET顯像藥物11C-α-DTBZ的純度和標記率,進一步影響最終的顯像效果。
目前,已報道的9-DM-α-DTBZ常用的制備方法為:α-二氫丁苯那嗪(α-DTBZ)在氫化鈉、六甲基磷酰三胺及N-甲基苯胺的作用下脫除甲基生成9-DM-α-DTBZ(Chirality,1997,9:59-62)。實驗表明,反應完成后,反應液中的溶劑和反應原料氫化鈉、六甲基磷酰三胺在反應后處理的過程中即可除去,而反應原料α-DTBZ及N-甲基苯胺則較難去除干凈。因此,在9-DM-α-DTBZ的制備過程中,可能混有的主要雜質為α-DTBZ及N-甲基苯胺。
到目前為止,還沒有關于9-DM-α-DTBZ及其雜質α-DTBZ和N-甲基苯胺的分離測定方法的相關報道。盡管有文獻報道:HPLC法測定丁苯那嗪原料藥含量,然而,一方面由于9-DM-α-DTBZ原料藥所含雜質(主要為α-DTBZ和N-甲基苯胺)和丁苯那嗪原料藥所含雜質的差別,另一方面由于9-DM-α-DTBZ和丁苯那嗪化學結構的差別,申請人通過研究發現,上述HPLC法測定丁苯那嗪原料藥含量并不能用于9-DM-α-DTBZ及其雜質的分離測定,采用此HPLC法時9-DM-α-DTBZ與雜質不能完全分離、分離度較差,進而導致9-DM-α-DTBZ及其雜質α-DTBZ和N-甲基苯胺的含量無法進行測定,也就無法對9-DM-α-DTBZ的純度和含量進行質量控制,難以確保PET顯像藥物11C-α-DTBZ的純度和標記率,從而不利于PET顯像藥物11C-α-DTBZ使用的有效性和安全性。
因此,建立一種9-去甲基-α-二氫丁苯那嗪及其雜質的分離測定方法具有重要的意義。
技術實現要素:
本發明要解決的第一個技術問題是現有技術無法實現9-去甲基-α-二氫丁苯那嗪原料藥中的9-DM-α-DTBZ及其主要雜質α-DTBZ和N-甲基苯胺完全分離的問題,本發明要解決的第二個技術問題是現有技術無法實現9-去甲基-α-二氫丁苯那嗪原料藥中的9-DM-α-DTBZ及其主要雜質α-DTBZ和N-甲基苯胺的含量同時進行測定的問題。
為解決上述技術問題,本發明提出一種9-去甲基-α-二氫丁苯那嗪及其雜質α-DTBZ和N-甲基苯胺的分離測定方法。
為解決上述技術問題,本發明是通過以下技術方案來實現的:
本發明提供一種9-去甲基-α-二氫丁苯那嗪及其雜質的分離測定方法,包括以下步驟:
稱取待測9-去甲基-α-二氫丁苯那嗪原料藥適量,加入體積比為40~90:10~60的醋酸銨緩沖液-甲醇進行溶解,制成濃度為0.5~1.5mg/mL的溶液,作為供試品溶液;
照高效液相色譜法,以反相色譜柱為色譜柱,以體積比為40~90:10~60的醋酸銨緩沖液-甲醇為流動相,流速為0.5~1.5mL/min,檢測波長為270~290nm,柱溫為20~35℃;
取所述供試品溶液5~15μL,注入高效液相色譜儀,測定。
優選地,本發明上述9-去甲基-α-二氫丁苯那嗪及其雜質的分離測定方法,以體積比為50~75:25~50的醋酸銨緩沖液-甲醇為流動相。
進一步優選地,本發明上述9-去甲基-α-二氫丁苯那嗪及其雜質的分離測定方法,
以體積比為70:30的醋酸銨緩沖液-甲醇為流動相;或者
以體積比為50:50的醋酸銨緩沖液-甲醇為流動相;或者
以體積比為75:25的醋酸銨緩沖液-甲醇為流動相。
進一步優選地,本發明上述9-去甲基-α-二氫丁苯那嗪及其雜質的分離測定方法,所述醋酸銨緩沖液的濃度為40~60mM、pH值為4~5。
進一步優選地,本發明上述9-去甲基-α-二氫丁苯那嗪及其雜質的分離測定方法,所述醋酸銨緩沖液的濃度為50mM、pH值為4.5。
進一步優選地,本發明上述9-去甲基-α-二氫丁苯那嗪及其雜質的分離測定方法,所述反相色譜柱的填充劑為十八烷基硅烷鍵合硅膠。
進一步優選地,本發明上述9-去甲基-α-二氫丁苯那嗪及其雜質的分離測定方法,所述反相色譜柱的填充劑為粒徑5μm的十八烷基硅烷鍵合硅膠。
進一步優選地,本發明上述9-去甲基-α-二氫丁苯那嗪及其雜質的分離測定方法,所述反相色譜柱為5μm、4.6×150mm的Sunfire C18反相色譜柱。
進一步優選地,本發明上述9-去甲基-α-二氫丁苯那嗪及其雜質的分離測定方法,包括以下步驟:
稱取待測9-去甲基-α-二氫丁苯那嗪原料藥適量,加入體積比為70:30的醋酸銨緩沖液-甲醇進行溶解,制成濃度為1mg/mL的溶液,作為供試品溶液;
照高效液相色譜法,以5μm、4.6×150mm的Sunfire C18反相色譜柱為色譜柱,以體積比為70:30的醋酸銨緩沖液-甲醇為流動相,流速為1mL/min,檢測波長為280nm,柱溫為25℃;所述醋酸銨緩沖液的濃度為50mM、pH值為4.5;
取所述供試品溶液10μL,注入高效液相色譜儀,測定;或者
稱取待測9-去甲基-α-二氫丁苯那嗪原料藥適量,加入體積比為50:50的醋酸銨緩沖液-甲醇進行溶解,制成濃度為1.0mg/mL的溶液,作為供試品溶液;
照高效液相色譜法,以5μm、4.6×150mm的Sunfire C18反相色譜柱為色譜柱,以體積比為50:50的醋酸銨緩沖液-甲醇為流動相,流速為1mL/min,檢測波長為280nm,柱溫為25℃;所述醋酸銨緩沖液的濃度為50mM、pH值為4.5;
取所述供試品溶液10μL,注入高效液相色譜儀,測定;或者
稱取待測9-去甲基-α-二氫丁苯那嗪原料藥適量,加入體積比為75:25的醋酸銨緩沖液-甲醇進行溶解,制成濃度為1mg/mL的溶液,作為供試品溶液;
照高效液相色譜法,以5μm、4.6×150mm的Sunfire C18反相色譜柱為色譜柱,以體積比為75:25的醋酸銨緩沖液-甲醇為流動相,流速為1mL/min,檢測波長為280nm,柱溫為25℃;所述醋酸銨緩沖液的濃度為50mM、pH值為4.5;
取所述供試品溶液10μL,注入高效液相色譜儀,測定。
進一步優選地,本發明上述9-去甲基-α-二氫丁苯那嗪及其雜質的分離測定方法,還包括如下9-去甲基-α-二氫丁苯那嗪對照品溶液、α-二氫丁苯那嗪對照品溶液、N-甲基苯胺對照品溶液的制備和測定的步驟:
取9-去甲基-α-二氫丁苯那嗪的對照品適量,制成濃度為0.5~1.5mg/mL的溶液,作為9-去甲基-α-二氫丁苯那嗪對照品溶液;
取α-二氫丁苯那嗪的對照品適量,制成濃度為0.5~1.5mg/mL的溶液,作為α-二氫丁苯那嗪對照品溶液;
取N-甲基苯胺的對照品適量,制成濃度為0.5~1.5mg/mL的溶液,作為N-甲基苯胺對照品溶液;
分別取所述9-去甲基-α-二氫丁苯那嗪對照品溶液、所述α-二氫丁苯那嗪對照品溶液和所述N-甲基苯胺對照品溶液各5~15μL,注入高效液相色譜儀,測定。
進一步優選地,本發明上述9-去甲基-α-二氫丁苯那嗪及其雜質的分離測定方法,還包括如下9-去甲基-α-二氫丁苯那嗪對照品溶液、α-二氫丁苯那嗪對照品溶液、N-甲基苯胺對照品溶液的制備和測定的步驟:
取9-去甲基-α-二氫丁苯那嗪的對照品適量,制成濃度為1mg/mL的溶液,作為9-去甲基-α-二氫丁苯那嗪對照品溶液;
取α-二氫丁苯那嗪的對照品適量,制成濃度為1mg/mL的溶液,作為α-二氫丁苯那嗪對照品溶液;
取N-甲基苯胺的對照品適量,制成濃度為1mg/mL的溶液,作為N-甲基苯胺對照品溶液;
分別取所述9-去甲基-α-二氫丁苯那嗪對照品溶液、所述α-二氫丁苯那嗪對照品溶液和所述N-甲基苯胺對照品溶液各10μL,注入高效液相色譜儀,測定。
與現有技術相比,本發明的上述技術方案具有以下優點:
本發明9-去甲基-α-二氫丁苯那嗪及其雜質的分離測定方法,以5μm、4.6×150mm的Sunfire C18反相色譜柱為色譜柱,以體積比為50~75:25~50的醋酸銨緩沖液-甲醇為流動相,不僅可以使9-去甲基-α-二氫丁苯那嗪原料藥中9-DM-α-DTBZ及其雜質α-DTBZ和N-甲基苯胺實現完全分離,而且可以對9-DM-α-DTBZ及其雜質α-DTBZ和N-甲基苯胺的含量同時進行測定,為9-去甲基-α-二氫丁苯那嗪原料藥中9-去甲基-α-二氫丁苯那嗪的含量測定和有關物質的含量測定提供了一種簡便、快速、靈敏的檢測方法,從而能夠實現對9-DM-α-DTBZ原料藥進行質量控制,進而確保PET顯像藥物11C-α-DTBZ的純度和標記率,有利于PET顯像藥物11C-α-DTBZ使用的有效性和安全性。
附圖說明
為了使本發明的內容更容易被清楚的理解,下面根據本發明的具體實施例并結合附圖,對本發明作進一步詳細的說明,其中:
圖1為實施例1中供試品溶液的HPLC色譜圖;
圖2為實施例1中9-去甲基-α-二氫丁苯那嗪對照品的HPLC色譜圖;
圖3為實施例1中α-二氫丁苯那嗪對照品的HPLC色譜圖;
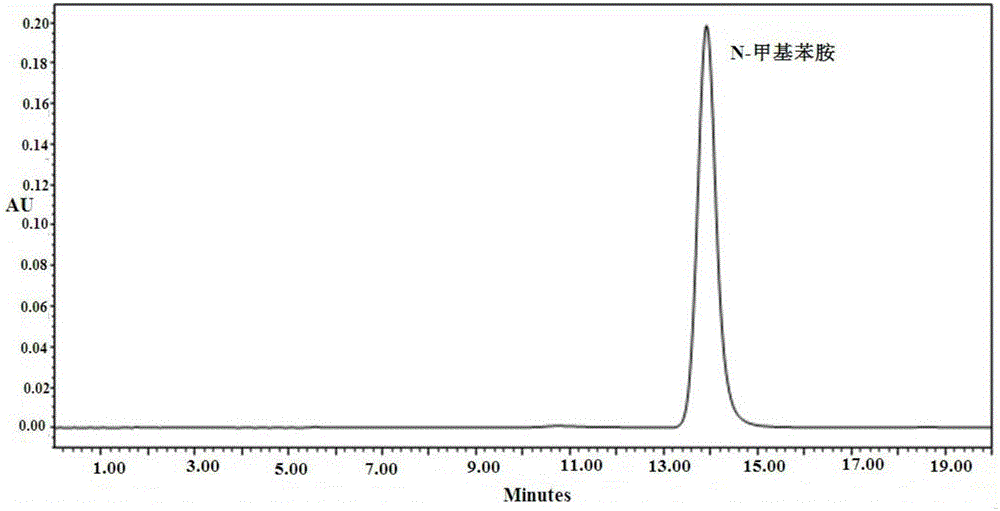
圖4為實施例1中N-甲基苯胺對照品的HPLC色譜圖;
圖5為實施例2中供試品溶液的HPLC色譜圖;
圖6為實施例3中供試品溶液的HPLC色譜圖;
圖7為對比例1中供試品溶液的HPLC色譜圖;
圖8為對比例2中供試品溶液的HPLC色譜圖;
圖9為對比例3中供試品溶液的HPLC色譜圖;
圖1-9中,9-DM-α-DTBZ表示的為9-去甲基-α-二氫丁苯那嗪,α-DTBZ表示的為α-二氫丁苯那嗪。
具體實施方式
本發明以下實施例和實驗例中,9-DM-α-DTBZ表示的為9-去甲基-α-二氫丁苯那嗪,α-DTBZ表示的為α-二氫丁苯那嗪。
實施例1
本實施例9-去甲基-α-二氫丁苯那嗪及其雜質的分離測定方法,包括以下步驟:
稱取待測9-去甲基-α-二氫丁苯那嗪原料藥適量,加入體積比為70:30的醋酸銨緩沖液(濃度為50mM、pH值為4.5)-甲醇進行溶解,制成濃度為1mg/mL的溶液,作為供試品溶液;
取9-去甲基-α-二氫丁苯那嗪的對照品適量,制成濃度為1mg/mL的溶液,作為9-去甲基-α-二氫丁苯那嗪對照品溶液;
取α-二氫丁苯那嗪的對照品適量,制成濃度為1mg/mL的溶液,作為α-二氫丁苯那嗪對照品溶液;
取N-甲基苯胺的對照品適量,制成濃度為1mg/mL的溶液,作為N-甲基苯胺對照品溶液;
照高效液相色譜法,以Sunfire C18反相色譜柱(5μm、4.6×150mm)為色譜柱,以體積比為70:30的醋酸銨緩沖液(濃度為50mM、pH值為4.5)-甲醇為流動相,流速為1mL/min,檢測波長為280nm,柱溫為25℃;
分別取供試品溶液、9-去甲基-α-二氫丁苯那嗪對照品溶液、α-二氫丁苯那嗪對照品溶液和N-甲基苯胺對照品溶液各10μL,注入高效液相色譜儀,測定。
本實施例中供試品溶液、9-去甲基-α-二氫丁苯那嗪對照品溶液、α-二氫丁苯那嗪對照品溶液和N-甲基苯胺對照品溶液的HPLC色譜圖分別如圖1-4所示。
由圖1-4可知,實施例1中,9-DM-α-DTBZ與反應原料α-DTBZ、N-甲基苯胺的保留時間差異大,三者完全分離,分離效果良好;9-DM-α-DTBZ的保留時間為6.22min,含量為65.1%;α-DTBZ的保留時間為10.60min,含量為16.6%;N-甲基苯胺的保留時間為13.88min,含量為18.3%。
實施例2
本實施例9-去甲基-α-二氫丁苯那嗪及其雜質的分離測定方法,包括以下步驟:
稱取待測9-去甲基-α-二氫丁苯那嗪原料藥適量,加入體積比為50:50的醋酸銨緩沖液(濃度為50mM、pH值為4.5)-甲醇進行溶解,制成濃度為1.0mg/mL的溶液,作為供試品溶液;
取9-去甲基-α-二氫丁苯那嗪的對照品適量,制成濃度為1mg/mL的溶液,作為9-去甲基-α-二氫丁苯那嗪對照品溶液;
取α-二氫丁苯那嗪的對照品適量,制成濃度為1mg/mL的溶液,作為α-二氫丁苯那嗪對照品溶液;
取N-甲基苯胺的對照品適量,制成濃度為1mg/mL的溶液,作為N-甲基苯胺對照品溶液;
照高效液相色譜法,以Sunfire C18反相色譜柱(5μm、4.6×150mm)為色譜柱,以體積比為50:50的醋酸銨緩沖液(濃度為50mM、pH值為4.5)-甲醇為流動相,流速為1mL/min,檢測波長為280nm,柱溫為25℃;
分別取供試品溶液、9-去甲基-α-二氫丁苯那嗪對照品溶液、α-二氫丁苯那嗪對照品溶液和N-甲基苯胺對照品溶液各10μL,注入高效液相色譜儀,測定。
本實施例中供試品溶液的HPLC色譜圖分別如圖5所示。
由圖5可知,實施例2中,9-DM-α-DTBZ與反應原料α-DTBZ分離效果稍差;9-DM-α-DTBZ的保留時間為2.45min,含量為65.4%;α-DTBZ的保留時間為3.02min,含量為15.4%;N-甲基苯胺的保留時間為6.68min,含量為19.2%。
實施例3
本實施例9-去甲基-α-二氫丁苯那嗪及其雜質的分離測定方法,包括以下步驟:
稱取待測9-去甲基-α-二氫丁苯那嗪原料藥適量,加入體積比為75:25的醋酸銨緩沖液(濃度為50mM、pH值為4.5)-甲醇進行溶解,制成濃度為1mg/mL的溶液,作為供試品溶液;
取9-去甲基-α-二氫丁苯那嗪的對照品適量,制成濃度為1mg/mL的溶液,作為9-去甲基-α-二氫丁苯那嗪對照品溶液;
取α-二氫丁苯那嗪的對照品適量,制成濃度為1mg/mL的溶液,作為α-二氫丁苯那嗪對照品溶液;
取N-甲基苯胺的對照品適量,制成濃度為1mg/mL的溶液,作為N-甲基苯胺對照品溶液;
照高效液相色譜法,以Sunfire C18反相色譜柱(5μm、4.6×150mm)為色譜柱,以體積比為75:25的醋酸銨緩沖液(濃度為50mM、pH值為4.5)-甲醇為流動相,流速為1mL/min,檢測波長為280nm,柱溫為25℃;
分別取供試品溶液、9-去甲基-α-二氫丁苯那嗪對照品溶液、α-二氫丁苯那嗪對照品溶液和N-甲基苯胺對照品溶液各10μL,注入高效液相色譜儀,測定。
本實施例中供試品溶液的HPLC色譜圖分別如圖6所示。
由圖6可知,實施例3中,9-DM-α-DTBZ與反應原料α-DTBZ、N-甲基苯胺的保留時間差異較大,三者完全分離,分離效果較好;9-DM-α-DTBZ的保留時間為9.65min,含量為66.2%;α-DTBZ的保留時間為16.28min,含量為16.7%;N-甲基苯胺的保留時間為18.79min,含量為17.1%。
實施例4
本實施例9-去甲基-α-二氫丁苯那嗪及其雜質的分離測定方法,包括以下步驟:
稱取待測9-去甲基-α-二氫丁苯那嗪原料藥適量,加入體積比為90:10的醋酸銨緩沖液(60mM、pH值為4)-甲醇進行溶解,制成濃度為1.5mg/mL的溶液,作為供試品溶液;
取9-去甲基-α-二氫丁苯那嗪的對照品適量,制成濃度為1mg/mL的溶液,作為9-去甲基-α-二氫丁苯那嗪對照品溶液;
取α-二氫丁苯那嗪的對照品適量,制成濃度為1mg/mL的溶液,作為α-二氫丁苯那嗪對照品溶液;
取N-甲基苯胺的對照品適量,制成濃度為1mg/mL的溶液,作為N-甲基苯胺對照品溶液;
照高效液相色譜法,以Sunfire C18反相色譜柱(5μm、4.6×150mm)為色譜柱,以體積比為90:10的醋酸銨緩沖液(60mM、pH值為4)-甲醇為流動相,流速為1.5mL/min,檢測波長為270nm,柱溫為35℃;
分別取供試品溶液、9-去甲基-α-二氫丁苯那嗪對照品溶液、α-二氫丁苯那嗪對照品溶液和N-甲基苯胺對照品溶液各5μL,注入高效液相色譜儀,測定。
實施例5
本實施例9-去甲基-α-二氫丁苯那嗪及其雜質的分離測定方法,包括以下步驟:
稱取待測9-去甲基-α-二氫丁苯那嗪原料藥適量,加入體積比為40:60的醋酸銨緩沖液(40mM、pH值為5)-甲醇進行溶解,制成濃度為0.5mg/mL的溶液,作為供試品溶液;
取9-去甲基-α-二氫丁苯那嗪的對照品適量,制成濃度為1mg/mL的溶液,作為9-去甲基-α-二氫丁苯那嗪對照品溶液;
取α-二氫丁苯那嗪的對照品適量,制成濃度為1mg/mL的溶液,作為α-二氫丁苯那嗪對照品溶液;
取N-甲基苯胺的對照品適量,制成濃度為1mg/mL的溶液,作為N-甲基苯胺對照品溶液;
照高效液相色譜法,以Sunfire C18反相色譜柱(5μm、4.6×150mm)為色譜柱,以體積比為40:60的醋酸銨緩沖液(40mM、pH值為5)-甲醇為流動相,流速為0.5mL/min,檢測波長為290nm,柱溫為20℃;
分別取供試品溶液、9-去甲基-α-二氫丁苯那嗪對照品溶液、α-二氫丁苯那嗪對照品溶液和N-甲基苯胺對照品溶液各15μL,注入高效液相色譜儀,測定。
實施例6
本實施例與實施例1的區別僅在于:流速為0.8mL/min;其余實驗條件和實驗操作步驟均與實施例1相同。
實施例7
本實施例與實施例1的區別僅在于:流速為1.2mL/min;其余實驗條件和實驗操作步驟均與實施例1相同。
對比例1
本對比例與實施例1的區別僅在于:制備供試品溶液的步驟中,加入體積比為20:80的醋酸銨緩沖液(濃度為50mM、pH值為4.5)-乙腈進行溶解;作為供試品溶液以體積比為20:80的醋酸銨緩沖液(濃度為50mM、pH值為4.5)-乙腈為流動相;以Lichrospher C18反相色譜柱(5μm、4.6×250mm)為色譜柱;流速為0.8mL/min;其余實驗條件和實驗操作步驟均與實施例1相同。
本對比例中供試品溶液的HPLC色譜圖分別如圖7所示。
由圖7可知,對比例1中,9-DM-α-DTBZ與反應原料α-DTBZ的保留時間基本一致,峰形幾乎重疊在一起,分離效果差;9-DM-α-DTBZ的保留時間為2.10min,α-DTBZ的保留時間為2.19min,N-甲基苯胺的保留時間為3.00min。
對比例2
本對比例與對比例1的區別僅在于:制備供試品溶液的步驟中,加入體積比為50:50的醋酸銨緩沖液(濃度為50mM、pH值為4.5)-乙腈進行溶解;作為供試品溶液以體積比為50:50的醋酸銨緩沖液(濃度為50mM、pH值為4.5)-乙腈為流動相;其余實驗條件和實驗操作步驟均與對比例1相同。
本對比例中供試品溶液的HPLC色譜圖分別如圖8所示。
由圖8可知,對比例2中,9-DM-α-DTBZ與反應原料α-DTBZ的分離度為0.7,分離效果差;9-DM-α-DTBZ的保留時間為2.24min,α-DTBZ的保留時間為1.73min,N-甲基苯胺的保留時間為6.93min。
對比例3
本對比例與對比例1的區別僅在于:制備供試品溶液的步驟中,加入體積比為80:20的醋酸銨緩沖液(濃度為50mM、pH值為4.5)-乙腈進行溶解;作為供試品溶液以體積比為80:20的醋酸銨緩沖液(濃度為50mM、pH值為4.5)-乙腈為流動相;其余實驗條件和實驗操作步驟均與對比例1相同。
本對比例中供試品溶液的HPLC色譜圖分別如圖9所示。
由圖9可知,對比例3中,9-DM-α-DTBZ與反應原料α-DTBZ的峰形完全重疊在一起,分離效果差;9-DM-α-DTBZ的保留時間為6.60min,α-DTBZ的保留時間為6.50min,N-甲基苯胺的保留時間為9.96min。
顯然,上述實施例僅僅是為清楚地說明所作的舉例,而并非對實施方式的限定。對于所屬領域的普通技術人員來說,在上述說明的基礎上還可以做出其它不同形式的變化或變動。這里無需也無法對所有的實施方式予以窮舉。而由此所引伸出的顯而易見的變化或變動仍處于本發明創造的保護范圍之中。
- 還沒有人留言評論。精彩留言會獲得點贊!