蛹蟲草提取物及其制備方法和應用與流程
本發明涉及天然活性產物
技術領域:
,特別是涉及一種蛹蟲草提取物及其制備方法和應用。
背景技術:
:糖尿病(Diabetesmellitus,DM)是一種以高血糖為特征的慢性代謝性疾病。近年來,社會不斷進步,人民生活水平不斷提高,糖尿病的發病率也不斷升高,糖尿病已經成為嚴重危害人類生命健康的惡性疾病之一,臨床上以高血糖為主要特點。我國目前糖尿病的總體患病率已達9.7%,同期糖尿病前期的患病率高達15.5%。根據調查結果所做出的推算顯示,我國總糖尿病患病人數達9千2百萬以上,糖尿病前期人數達1億4千8百萬以上。近年來的研究發現,蛋白酪氨酸磷酸酶(ProteinTyrosinePhosphatase,PTP)與2型糖尿病的胰島素信號傳導、高血脂、肥胖密切相關。PTPs家族可分為8個亞族:①酪氨酸特異性PTPs,②雙特異性PTPs,③細胞分裂周期蛋白25(cdc25)亞族,④第10號染色體缺失的磷酸酶和張力蛋白同源基因(PTEN)亞族,⑤Myotubularins,⑥再生肝磷酸酶(PRL)亞族,⑦低分子量PTPs,⑧新發現的cdc14亞族。其中,與胰島素信號調節緊密相關的是酪氨酸特異性PTPs。PTP1B是胞內PTPs的代表,是第一個被鑒定并純化的哺乳動物PTP,廣泛表達于肌肉、心、肝、腎、腦等各種組織中。PTP-1B在胰島素抵抗中發揮了重要的作用,是胰島素信號轉導中一個十分關鍵的分子,可以負性調節胰島素和瘦素的信號轉導。胰島素信號傳導首先需要胰島素與胰島素受體結合。胰島素受體是由α2β2亞單位組成的四聚體,α亞單位與胰島素結合后β亞單位的酪氨酸及胰島素受體底物發生磷酸化而被激活。磷脂酰肌醇3激酶(phosphatidylinositol3-kianse,PI3K)等下游蛋白也被活化,葡萄糖轉運蛋白4由細胞內轉至細胞膜,最終葡萄糖進入細胞。在上述過程中的磷酸化反應可以被PTP-1B等因子逆轉,從而阻止胰島素信號的傳導。如果PTP-1B結構或活性改變,將改變其對胰島素信號傳導的抑制作用。-外周組織胰島素抵抗和胰島β細胞功能受損是2型糖尿病發病機制的兩個基本條件,而胰島素信號傳導被抑制是胰島素抵抗的重要環節。PTP-1B升高表達可引起胰島素抵抗,從而引發2型糖尿病。有資料表明,PTP-1B的表達水平在發生糖尿病或胰島素抵抗時明顯升高。使轉基因鼠表達人PTP-1B的有關研究中發現,與對照組相比,實驗組鼠肌肉中胰島素刺激引起的胰島素受體酪氨酸磷酸化水平降低35%,PI3K活性降低40%~60%,葡萄糖轉運所需的蛋白激酶c活性也減弱;表達PTP-1B的小鼠全身葡萄糖清除和肌肉的葡萄糖攝取減少了40%~50%。并有研究表明體重指數正常的初診2型糖尿病較正常對照者存在明顯的胰島素抵抗,而PTP-1B在前者內臟脂肪組織中的表達較后者顯著增高,幾乎是后者的4倍。可見內臟脂肪組織PTP-1B的表達升高在胰島素抵抗中也有重要作用。另有研究報道采用PTP-1B反義寡核苷酸處理糖尿病模型小鼠(ob/ob小鼠),抑制其PTP-1B表達,發現在肝、脂肪和骨骼肌內PTP-1B的表達下調時,ob/ob小鼠血糖恢復正常,與糖代謝有關的各個指標也都趨于正常。以上結果說明,PTP-1B在拮抗胰島素信號傳導中具有重要作用。糖尿病患者體內PTP,尤其是PTP-1B的含量及活性的異常可能存在多方面的原因,基因變異被認為是主要因素。有研究表明,先用限速酶AVAI對653個樣本的一種單核苷酸多肽鏈消化,再利用外顯子PCR放大技術對其編碼發現,帶有PTP981T/981C基因型的人群患2型糖尿病的概率大大增加。目前,PTP1B抑制劑很多,但是大多都是人工合成得到的,具有很大的副作用,臨床上成藥性很差,到目前為止,還沒有一個PTP1B抑制劑在臨床上進行應用。而中草藥具有天然、多效、毒性小等特點。從中藥和天然藥物中尋找能夠抑制PTP1B的有效成分是迫切需要解決的課題。技術實現要素:基于此,有必要針對上述問題,提供一種蛹蟲草提取物,該蛹蟲草提取物具有較好的PTP1B抑制作用,為臨床上開發降血糖、防治胰島素抵抗、糖尿病的藥物提供一個新的途徑。一種蛹蟲草提取物的制備方法,其特征在于,包括以下步驟:制備醇提物:取蛹蟲草,加入體積百分濃度為80-100%的乙醇水溶液作為提取溶劑,加熱提取,然后去除固體不溶物,得醇提液,回收所述醇提液中的溶劑,得醇提物,即為蛹蟲草提取物。可以理解的,所述去除固體不溶物的方式有多種,如常規技術中的過濾,離心等手段均可。蛹蟲草(CordycepsmilitarisL.Link),又名北冬蟲夏草、北蟲草,屬子囊菌門(Ascomycota),糞殼菌綱(Sordariomycetes),肉座菌目(Hypocreales),蟲草菌科(Cordycipitaceae),蟲草屬(Cordyceps)。目前世界上已發現蟲草屬真菌350多種,其中我國記錄有61種。《新華本草綱要》記載其“性平,味甘,有益肺腎,補精髓,止血化痰”,用于肺結核、老人虛弱和貧血等疾病的治療。本發明人經過長期的實驗研究后發現,采用上述方法制備得到的蛹蟲草提取物,對蛋白酪氨酸磷酸酶1B(PTP1B)的抑制作用,可用于開發制備降血糖、防治胰島素抵抗、糖尿病的藥物中。在其中一個實施例中,所述制備醇提物步驟中,每次按照每克蛹蟲草加入10mL-20mL提取溶劑的量加入體積百分濃度為90%-100%的乙醇水溶液,加熱提取1次-5次,每次30min-120min。在其中一個實施例中,所述蛹蟲草提取物的制備方法還包括以下步驟:制備環己烷溶解物:取所述醇提物,加入環己烷,攪拌使所述醇提物充分溶解,然后分離固體不溶物與溶液,分別得環己烷殘渣和環己烷溶液,回收所述環己烷溶液中的溶劑,得環己烷溶解物;制備乙酸乙酯溶解物:取所述環己烷殘渣,加入乙酸乙酯,攪拌使所述環己烷殘渣充分溶解,然后分離固體不溶物與溶液,分別得乙酸乙酯殘渣和乙酸乙酯溶液,回收所述乙酸乙酯溶液中的溶劑,得乙酸乙酯溶解物;制備乙酸乙酯萃取物:取所述乙酸乙酯殘渣,加入乙醇,攪拌使所述乙酸乙酯殘渣充分溶解,然后分離固體不溶物與溶液,得到乙醇溶液,回收所述乙醇溶液中的溶劑,得乙醇溶解物;將所述乙醇溶解物加水溶解,隨后再加入乙酸乙酯萃取,取乙酸乙酯層,回收溶劑后得到乙酸乙酯萃取物;所述環己烷溶解物、乙酸乙酯溶解物和乙酸乙酯萃取物中的至少一種即為所述蛹蟲草提取物。在其中一個實施例中,所述制備環己烷溶解物步驟中,每次按照每克醇提物加入50mL-100mL環己烷的量加入環己烷,攪拌使所述醇提物充分溶解2次-6次;所述制備乙酸乙酯溶解物步驟中,每次按照每克環己烷殘渣加入50mL-100mL乙酸乙酯的量加入乙酸乙酯,攪拌使所述環己烷殘渣充分溶解2次-6次;所述制備乙酸乙酯萃取物步驟中,按照每克乙醇溶解物加入10mL-60mL水的量加入水溶解得水溶液,隨后每次按照0.5-3倍水溶液體積的量加入乙酸乙酯,萃取2次-5次。在其中一個實施例中,所述蛹蟲草提取物的制備方法還包括以下步驟:取所述環己烷溶解物,以正相硅膠色譜柱為固定相,以體積百分比為200∶1~10∶1的環己烷-乙酸乙酯混合溶液為流動相進行洗脫,以TLC薄層色譜檢測洗脫液,合并相同組份,分離得到組份3、組份4、組份5、組份6、組份7,所述組份3、組份4、組份5、組份6、組份7的極性依次由小至大;所述TLC薄層色譜檢測的條件為:固定相為正相硅膠,展開劑分別為170∶1和80∶1的環己烷-乙酸乙酯混合溶液,顯色劑為10%的硫酸水溶液,于日光下檢視。所述組份3、組份4、組份5、乙酸乙酯溶解物和乙酸乙酯萃取物中的至少一種即為所述蛹蟲草提取物。在其中一個實施例中,所述蛹蟲草提取物的制備方法還包括以下步驟:取所述組份4,以反相C18硅膠色譜柱進行分離純化,具體條件如下:固定相:C18反相硅膠柱,柱直徑為7.8mm,柱長度為100mm;流動相:甲醇-水梯度洗脫,具體為:0至15min,水相體積百分數由80%降至50%,15至30min,水相體積百分數由50%降至15%,30至90min,水相體積百分數由15%降至0%;流速:3mL/min;檢測波長:190nm和/或254nm;收集第66min和第69min色譜峰的洗脫液,回收溶劑,分別得到單體化合物1和單體化合物2;所述單體化合物1的結構如下:所述單體化合物2的結構如下:所述組份3、單體化合物1、單體化合物2、組份5、乙酸乙酯溶解物和乙酸乙酯萃取物中的至少一種即為所述蛹蟲草提取物。本發明還公開了上述的蛹蟲草提取物的制備方法制備得到的蛹蟲草提取物。本發明還公開了上述的單體化合物1和/或單體化合物2。本發明還公開了上述的蛹蟲草提取物、單體化合物1和單體化合物2中的至少一種在作為抑制蛋白酪氨酸磷酸酶1B的抑制劑和/或作為胰島素增敏劑中的應用。在其中一個實施例中,所述蛋白酪氨酸磷酸酶1B的抑制劑,和/或所述胰島素增敏劑在制備用于預防和治療糖尿病、肥胖癥及其并發癥的保健食品或藥物中應用。在其中一個實施例中,所述藥物或保健食品的劑型為片劑、顆粒劑、硬膠囊、軟膠囊、口服液、丸劑或滴丸。上述劑型可以采用本領域技術人員熟知的方法來進行藥劑的制備。根據需要,可以加入各種藥學上可接受的載體。所述的載體包括藥學領域常規的稀釋劑、賦形劑、填充劑、粘合劑、濕潤劑、崩解劑、吸收促進劑、表面活性劑、吸附載體、潤滑劑等。本發明的藥劑在制備時,可選擇的填充劑包括但不限于:淀粉、糖粉、磷酸鈣、糊精、微晶纖維素、乳糖、預膠化淀粉、甘露醇等;可選擇的粘合劑包括但不限于:羧甲基纖維素鈉、PVP-K30、羥丙基纖維素、淀粉漿、甲基纖維素、乙基纖維素、羥丙基甲基纖維素、膠化淀粉等;可選擇的崩解劑包括但不限于:干淀粉、交聯聚維酮、交聯羧甲基纖維素鈉、羧甲基淀粉鈉、低取代羥丙基纖維素等;可選擇的潤滑劑包括但不限于:硬脂酸鎂、滑石粉、十二烷基硫酸鈉、微粉硅膠等。與現有技術相比,本發明具有以下有益效果:本發明的一種蛹蟲草提取物,對蛹蟲草原料進行醇提,得到的醇提物即為蛹蟲草提取物具有蛋白酪氨酸磷酸酶1B(PTP1B)的抑制作用,可用于開發制備降血糖、防治胰島素抵抗、糖尿病的藥物中。并且,蛹蟲草藥食同源,按照上述方法制備的醇提物從蛹蟲草中直接提取,具有毒性低、安全性高、可長期服用的優點。進一步的,將上述醇提物進行分離純化,得到環己烷溶解物、乙酸乙酯溶解物、乙酸乙酯萃取物等有效部位,其PTP1B抑制活性優于上述醇提物。更進一步來說,還可將上述環己烷溶解物進行分離純化,得到組份3、組份4、組份5及單體化合物1、單體化合物2,具有更佳的PTP1B抑制活性。特別是其中的單體化合物1和單體化合物2對PTP1B的半數抑制濃度分別為21.3μM和19.7μM。為臨床上開發降血糖、防治胰島素抵抗、糖尿病的藥物提供一個新的途徑。附圖說明圖1為實施例1制備得到的蛹蟲草提取物對PTP1B的抑制率柱形圖;圖2為實施例2制備得到的蛹蟲草提取物對PTP1B的抑制率柱形圖。具體實施方式為了便于理解本發明,下面將參照相關附圖對本發明進行更全面的描述。附圖中給出了本發明的較佳實施例。但是,本發明可以以許多不同的形式來實現,并不限于本文所描述的實施例。相反地,提供這些實施例的目的是使對本發明的公開內容的理解更加透徹全面。除非另有定義,本文所使用的所有的技術和科學術語與屬于本發明的
技術領域:
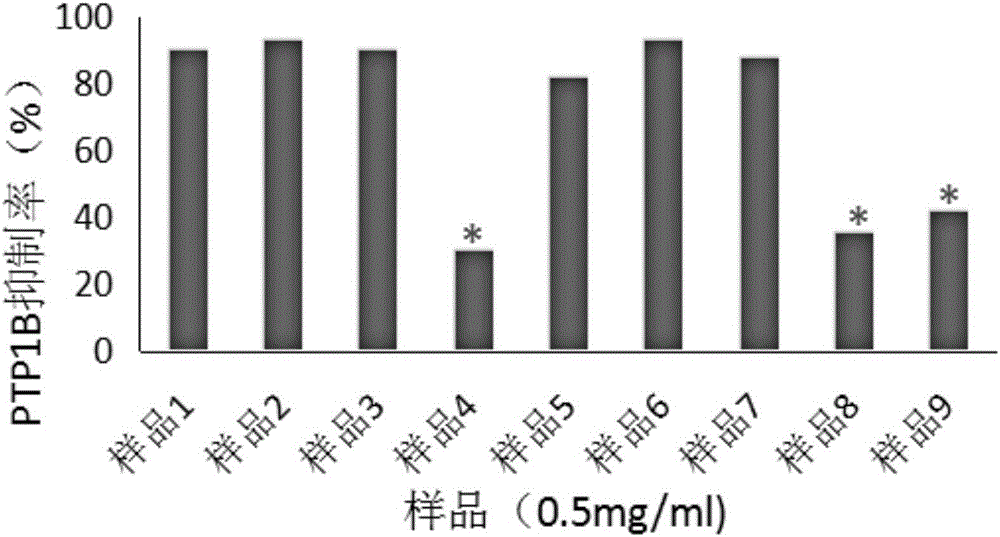
的技術人員通常理解的含義相同。本文中在本發明的說明書中所使用的術語只是為了描述具體的實施例的目的,不是旨在于限制本發明。本文所使用的術語“和/或”包括一個或多個相關的所列項目的任意的和所有的組合。實施例1蛹蟲草提取物,通過以下方法制備得到:一、制備醇提物。取蛹蟲草粗粉200g,按固液比1∶10(每克原料固體加入10ml溶液)加入2000ml體積百分濃度為95%的乙醇水溶液,80℃水浴回流2h,共4次,過濾,得醇提液,合并醇提液;減壓濃縮回收乙醇,合并濃縮液,繼續于敞口容器中55℃加熱烘干,即得醇提物,稱重為35.4g。二、制備環己烷溶解物。將上述醇提物置于較大容器中,加入1770ml(50倍)環己烷,攪拌,靜止,過濾,收集環己烷溶解液,此過程重復進行4次,環己烷溶解液減壓濃縮回收環己烷,合并濃縮液,繼續于敞口容器中55℃加熱揮盡環己烷,即為環己烷溶解物,稱重為5.6g。三、制備乙酸乙酯溶解物。將上述環己烷溶解后的殘渣不溶物用乙酸乙酯作為溶解溶劑,按照步驟二中制備環己烷溶解物的方法操作,制備得到乙酸乙酯溶解物,稱重為2.7g。四、制備正丁醇溶解物。將上述乙酸乙酯溶解后的殘渣不溶物用正丁醇作為溶解溶劑,按照步驟二中制備環己烷溶解物的方法操作,制備得到正丁醇溶解物,稱重20.8g。五、正相硅膠分離純化。取上述步驟二制備得到的環己烷溶解物,采用正向硅膠色譜柱分離,環己烷∶乙酸乙酯(200∶1~10∶1)為流動相梯度洗脫,TLC薄層色譜鑒定相同組分合并。其中,TLC薄層色譜檢測的條件為:固定相為正相硅膠,展開劑分別為170∶1和80∶1的環己烷-乙酸乙酯混合溶液,顯色劑為10%的硫酸水溶液,于日光下檢視,將TLC薄層色譜鑒定相同組分合并后,得到極性依次由小至大的組份3、組份4、組份5、組份6、組份7,稱重分別為0.58g、2.12g、1.68g、0.65g、0.45g。六、反向C18硅膠色譜柱分離純化。取步驟五制備得到的組份4(2.12g),用甲醇溶解,0.2微米微孔濾膜過濾,隨后用半制備HPLC(日本島津)分離純化,半制備柱為WatersC18柱(柱型號為186001155,填料粒徑為5μm,規格為7.8mm×100mm),甲醇水梯度洗脫。具體梯度為:0~15min,水80%~50%,15~30min,水50%~15%,30~90min,水15%~0%,90~120min,水0%;檢測波長190nm,254nm;流速3mL/min。分別收集于66min,69min出現色譜峰的洗脫液,回收溶劑后得到單體化合物1和單體化合物2。經FABMS,1H-NMR,13C-NMR,COSY,HMBC,HMQC等波譜數據鑒別后確認該單體化合物1的化學式為:C48H89NO8;結構式如下:上述化合物的表征數據為:FABMS(negative)m/z807[M]+;1H-NMR(500MHz,CD3OD):δ5.81(1H,dt,J=15.1,6.7Hz),5.70(1H,dt,J=15.5,5.5Hz),5.46(1H,dd,J=15.1,6.1Hz),5.44(1H,dt,J=15.5,6.8Hz),5.12(1H,t,J=5.5Hz),4.41(1H,d,J=6.1Hz),4.24(1H,d,J=7.8Hz),4.12(1H,dd,J=10.4,5.4Hz),3.92(1H,m),3.69(1H,dd,J=10.4,3.5Hz),3.22-3.65(56H,m),2.01-2.12(10H,m),1.98(2H,t,J=7.6Hz),1.59(3H,s),1.28-1.34(44H,m),0.88(9H,t,J=6.9Hz).經FABMS,1H-NMR,13C-NMR,COSY,HMBC,HMQC等波譜數據鑒別后確認該單體化合物2的化學式為:C41H77NO9;結構式如下:上述化合物的表征數據為:FABMS(negative)m/z727[M]+;1H-NMR(500MHz,CD3OD):δ5.75(1H,dt,J=15.4,5.4Hz),5.50(1H,dd,J=15.4,6.8Hz),5.08(1H,t,J=7.0Hz),4.30(1H,d,J=7.8Hz),4.11(1H,t,J=6.8Hz),4.06(1H,dd,J=10.2,5.6Hz),4.00(1H,m),3.73(1H,dd,J=10.2,3.6Hz),3.33(2H,m),3.17-3.23(5H,m),2.07(4H,m),1.99(2H,m),1.57(3H,s),1.36(2H,m),1.30-1.39(-CH2-),0.94(6H,t,J=6.3Hz).實施例2蛹蟲草提取物,通過以下方法制備得到:一、制備醇提物。取蛹蟲草粗粉500g,按固液比1∶15(每克原料固體加入15ml溶液)加入7500ml的無水乙醇,80℃水浴回流1.5h,共4次,過濾,得醇提液,合并醇提液;減壓濃縮回收乙醇,合并濃縮液,繼續于敞口容器中55℃加熱烘干,即得醇提物,稱重為90g。二、制備環己烷溶解物。將上述醇提物置于較大容器中,加入5.4L(60倍)環己烷,攪拌,靜止,過濾,收集環己烷溶解液,此過程重復進行5次,環己烷溶解液減壓濃縮回收環己烷,合并濃縮液,繼續于敞口容器中55℃加熱揮盡環己烷,即為環己烷溶解物,稱重為13.8g。三、制備乙酸乙酯溶解物。將上述環己烷溶解后的殘渣不溶物用乙酸乙酯作為溶解溶劑,按照步驟二中制備環己烷溶解物的方法操作,制備得到乙酸乙酯溶解物,稱重為6.8g。四、制備無水醇溶解物。將上述乙酸乙酯溶解后的殘渣不溶物用無水乙醇作為溶解溶劑,按照步驟二中制備環己烷溶解物的方法操作,制備得到無水乙醇溶解物,稱重49.6g。五、制備乙酸乙酯萃取物。將上述無水乙醇溶解后的殘渣不溶物加1000ml水溶解,加入2倍量的乙酸乙酯萃取5次,合并乙酸乙酯萃取液,采用減壓濃縮方法,得到乙酸乙酯萃取物16.2g,與乙酸乙酯溶解物合并,為組份1,共為23g。六、制備正丁醇萃取物。將余下的水層在60℃加熱揮盡乙酸乙酯,水層按照上述步驟中的方法用正丁醇萃取,采用減壓濃縮方法,得正丁醇萃取物27.3g,為組份2。七、正相硅膠分離純化。按照實施例1中正相硅膠分離純化的方法,取上述步驟二制備得到的環己烷溶解物進行分離純化,得到與實施例1一致的組份3、組份4、組份5、組份6、組份7,稱重分別為1.2g、5.1g、3.9g、2.0g、1.1g。八、反向C18硅膠色譜柱分離純化。取步驟七制備得到的組份4(5.1g),按照實施例1中反向C18硅膠色譜柱分離純化的方法操作,同樣得到單體化合物1和單體化合物2。實驗例1取實施例1制備得到的乙酸乙酯溶解物、正丁醇醇溶解物、組份3、組份4、組份5、組份6、組份7、單體化合物1和單體化合物2,測試各成分對蛋白酪氨酸磷酸酶1B的抑制作用。一、材料1、取乙酸乙酯溶解物、正丁醇溶解物、組份3、組份4、組份5、組份6、組份7,均配成濃度為0.5mg/mL;2、單體化合物1和單體化合物2;3、PTP1B,購自Prospec公司;4、Tris,購自有限責任公司背景鼎國生物技術有限責任公司;5、HCl,購自西隴化工股份有限公司;6、P-NPP,購自北京鼎國昌盛生物技術有限公司;7、熊果酸、科羅索酸,均購自購自北京鼎國昌盛生物技術有限公司,均配成濃度為0.5mg/mL;8、其余試劑均為國產試劑分析純;二、方法參照文獻(Zhang,Y.L.,etal.″Geranylated2-arylbenzofuransfromMorusalbavar.tataricaandtheiralpha-glucosidaseandproteintyrosinephosphatase1Binhibitoryactivities.″Fitoterapia92:116-126.(2014))報道的方法測定PTP1B的活性。簡述如下:PTP1B活性測定反應體系,包括:100mMp-NPP(底物),10μL;0.01μg/μL的PTP1B,20μL;緩沖液(25mMTris-HCl,1mMEDTA,1mMDTT,2mMβ-巰基乙醇;pH7.5),65μL;樣品5μL,總反應體系100μL。測定方法:在96孔板中先加入65μL緩沖液,再分別加入5μL樣品,后加入20μL0.01μg/μL的PTP1B酶,30℃孵育5min,后加入10μL100mMp-NPP啟動反應,30℃反應30min,用50μL預冷的3MNaOH終止反應,在405nm處測定吸光度值(OD)。按照上述酶活性測定方法,將表1所示的不同樣品用DMSO溶解,405nm處測定吸光度值,每個濃度設置三個復孔,按照如下公式計算抑制率:抑制率=(OD陰性對照組-OD空白對照組)-(OD樣品-OD樣品空白對照組)/(OD陰性對照組-OD空白對照組)×100%其中:OD陰性對照組包括:緩沖液+PTP1B酶+底物;OD空白對照組包括:緩沖液+底物;OD樣品包括:緩沖液+樣品+PTP1B酶+底物;OD樣品空白對照組包括:緩沖液+樣品+底物;三、實驗結果1、乙酸乙酯溶解物、正丁醇溶解物、組份3、組份4、組份5、組份6、組份7的抑制活性結果如下表和圖1所示:表1不同樣品對PTP1B的抑制作用*表示分別與樣品1和樣品2相比,均表現為具有統計學差異P<0.05。從上述結果中可以看出,在與陽性藥熊果酸和科羅索酸在相同濃度(均為0.5mg/mL)下,乙酸乙酯溶解物、組份3、組份4和組份5對蛋白酪氨酸磷酸酶1B(PTP1B)具有較強的抑制作用;正丁醇溶解物和組份6,7對蛋白酪氨酸磷酸酶1B(PTP1B)的抑制作用較弱(與陽性藥熊果酸和科羅索酸相比,有顯著性差異P<0.05)。2、參照上述方法,測定單體化合物1和單體化合物2的IC50,結果如下表所示。表2.單體化合物對PTP1B的抑制作用從上述結果中可以看出,當單體化合物1的濃度為21.3μM時,對PTP1B有50%抑制作用(IC50);當單體化合物2的濃度為19.7μM時,對PTP1B有50%抑制作用(IC50)。其抑制作用均比組份4強,也就是說,單體化合物1和單體化合物2經過分離純化后,去除了組份4中無活性或低活性成分,具有最佳的PTP1B抑制作用,且具有干擾成分少,副作用低的優點。實驗例2取實施例2制備得到的組份1、組份2、組份3、組份4、組份5、組份6、組份7、單體化合物1和單體化合物2,測試各成分對蛋白酪氨酸磷酸酶1B的抑制作用。一、材料同實驗例1。二、方法同實驗例1。三、實驗結果1、組份1、組份2、組份3、組份4、組份5、組份6、組份7的抑制活性結果如下表和圖2所示:表3不同樣品對PTP1B的抑制作用成分濃度(mg/mL)抑制率(%)樣品1熊果酸0.590.1樣品2科羅索酸0.592.9樣品3組份10.589.8樣品4組份20.530.1*樣品5組份30.581.4樣品6組份40.592.7樣品7組份50.587.8樣品8組份60.535.2*樣品9組份70.541.9**表示分別與樣品1和樣品2相比,均表現為具有統計學差異P<0.05。從上述結果中可以看出,在與陽性藥熊果酸和科羅索酸相同濃度(均為0.5mg/mL)下,組份1、組份3、組份4和組份5對蛋白酪氨酸磷酸酶1B(PTP1B)具有較強的抑制作用;組份2,6,7對蛋白酪氨酸磷酸酶1B(PTP1B)的抑制作用較弱,(與陽性藥熊果酸和科羅索酸相比,有顯著性差異P<0.05)。2、參照上述方法,測定單體化合物1和單體化合物2的IC50,結果如下表所示。表4.單體化合物對PTP1B的抑制作用從上述結果中可以看出,當單體化合物1的濃度為21.1μM時,對PTP1B有50%抑制作用(IC50);當單體化合物2的濃度為19.8μM時,對PTP1B有50%抑制作用(IC50)。其抑制作用均比組份4強,也就是說,單體化合物1和單體化合物2經過分離純化后,去除了組份4中無活性或低活性成分,具有最佳的PTP1B抑制作用,且具有干擾成分少,副作用低的優點。以上所述實施例的各技術特征可以進行任意的組合,為使描述簡潔,未對上述實施例中的各個技術特征所有可能的組合都進行描述,然而,只要這些技術特征的組合不存在矛盾,都應當認為是本說明書記載的范圍。以上所述實施例僅表達了本發明的幾種實施方式,其描述較為具體和詳細,但并不能因此而理解為對發明專利范圍的限制。應當指出的是,對于本領域的普通技術人員來說,在不脫離本發明構思的前提下,還可以做出若干變形和改進,這些都屬于本發明的保護范圍。因此,本發明專利的保護范圍應以所附權利要求為準。當前第1頁1 2 3
相關技術
網友詢問留言
已有0條留言
- 還沒有人留言評論。精彩留言會獲得點贊!
1