一種基于小麥自然群體全基因組關聯分析的赤霉病抗性關聯區間Meta分析方法和應用
本發明屬于數據分析,具體涉及一種基于小麥自然群體全基因組關聯分析的赤霉病抗性關聯區間meta分析方法和應用。
背景技術:
1、小麥(triticum?aestivum?l.)作為全球主要的糧食作物之一,其產量和品質對全球糧食安全具有重要影響。然而,小麥的生產過程中常受多種病害的侵襲,赤霉病(fusarium?head?blight,fhb)就是其中最具破壞性的一種。赤霉病不僅導致小麥產量顯著下降,還會產生真菌毒素(如嘔吐毒素),危害人畜健康。因此,培育抗赤霉病小麥品種是小麥育種研究的重要目標之一。
2、小麥抗病育種的核心在于準確識別與利用抗病性狀相關的數量性狀基因座(qtl)。傳統的qtl作圖方法依賴兩個親本衍生的遺傳群體,該類群體由于受限于雙親本遺傳背景和雙親多態性位點少的限制,研究結果在不同群體間常存在較大差異。為解決這一問題,全基因組關聯分析(gwas)近年來得到了廣泛應用,其能夠在眾多遺傳背景不同的育種群體中高效篩選與目標性狀相關的遺傳變異。然而,gwas結果也常因群體來源、大小、實驗條件和環境的不同而存在不一致性,導致研究結果難以比較和利用。
3、為了提高關聯區間定位結果的準確性和穩定性,meta分析成為一種有效的解決方案。meta分析通過整合已發表的研究結果,從而提高qtl定位結果的可信度。這種方法已被廣泛應用于多種作物的雙親本衍生的遺傳群體,但在作物(包括小麥)自然群體中尚未有應用的先例,主要原因是gwas分析結果給出的是關聯分子標記位點,而非區間。
4、cn113113086a公開了一種小麥抗赤霉病dsmqtl分析方法,但該方法中赤霉病(fhb)抗性位點的鑒定主要集中于兩個親本衍生獲得的特定小麥遺傳群體,其范圍受限于特定的極少數品種,由于兩個親本的dna差異性有限,遺傳背景狹窄,導致無法發現更多的穩定遺傳位點。
技術實現思路
1、針對現有技術的不足,本發明提供一種基于小麥自然群體全基因組關聯分析的赤霉病抗性關聯區間meta分析方法和應用,通過meta分析整合多篇赤霉病抗性全基因組關聯分析的結果,篩選和挖掘不同于雙親本作圖群體中已報道的與赤霉病抗性穩定關聯的新的關聯區間,為赤霉病抗性改良提供給新的靶標位點,為赤霉病抗性遺傳研究和精準育種提供重要信息。
2、本發明是通過以下技術方案實現的:
3、一種基于小麥自然群體全基因組關聯分析的赤霉病抗性關聯區間meta分析方法,包括以下步驟:
4、步驟1)基于關鍵檢索詞在數據知識服務平臺中進行檢索以篩選出對應的文獻,所述關鍵檢索詞為:小麥、赤霉病、全基因組關聯分析;
5、步驟2)對篩選得到的文獻進行收錄,對收錄文獻的標題、年份、snp名稱、關聯區間左側位置、關聯區間右側位置、關聯區間peak位置、snp?p?value以及pve值進行整理并匯總出原始關聯區間;對于原始關聯區間一側物理位置不明確或者只知道snp位置的,利用小麥連鎖不平衡衰減距離衍生出關聯區間位置;根據小麥品種中國春測序圖譜iwgsc?refseqv2.1,對整理好的數據進行物理位置轉換;
6、步驟3)將整理好的數據導入ammqtl軟件中,并采用最小二乘法構建模型,得到model1-ammqtl、model2-ammqtl、model3-ammqtl、model4-ammqtl這4個模型,每條染色體上會有1、2、3、4個ammqtl;
7、步驟4)對4個模型分別進行擬合檢驗,選擇最符合qtl可靠性積累曲線的模型,并對最終輸出meta?qtl進行篩選,得到meta分析結果。
8、優選地,步驟1)所述文獻篩選的標準為:①小麥群體只選用自然群體;②小麥赤霉病抗性性狀只關注小麥抗擴展以及抗毒素積累;③在gwas文章中只關注snp的結果;④選用重現性snp關聯結果。
9、優選地,步驟1)所述文獻為11篇國內外公開發表的關于小麥赤霉病自然群體gwas研究的文獻,不包括連鎖分析和qtl驗證的文獻。
10、優選地,步驟4)所述篩選的標準為:①qtl置信度的積累曲線聚類成峰,模型可以擬合;②一個ammqtl至少有2個完全或部分重疊的原始物理區間。
11、優選地,步驟4)所述meta分析結果包括64個meta關聯區間,其中35個是與小麥抗赤霉病關聯的新的穩定meta關聯區間,其余29個與小麥雙親本衍生的遺傳群體meta研究結果一致。
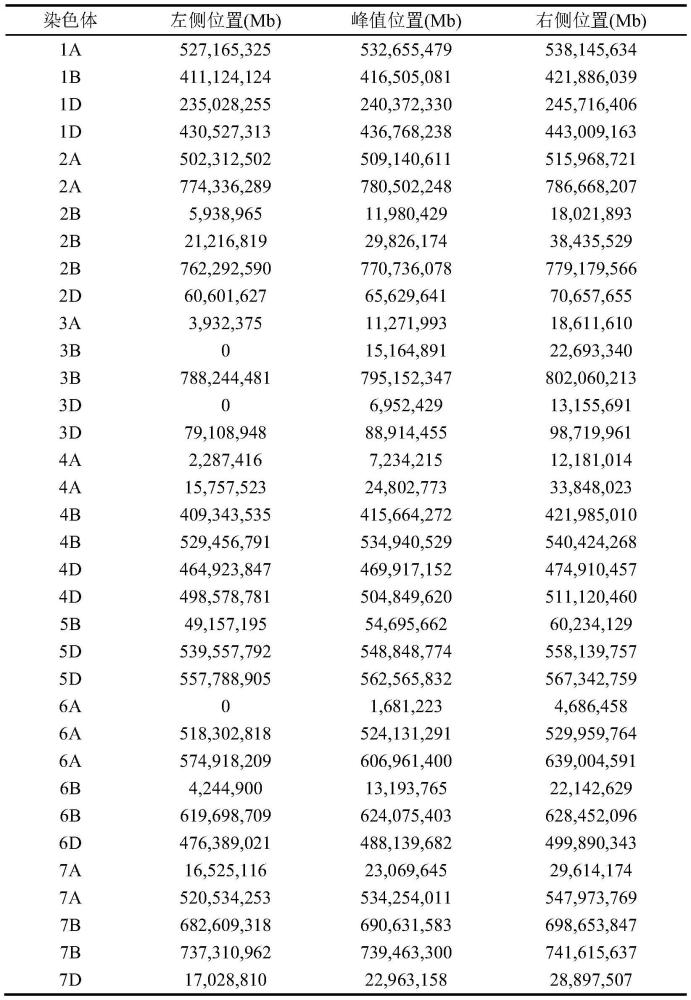
12、優選地,所述35個與小麥抗赤霉病關聯的新的穩定meta關聯區間的信息如下:
13、
14、上述基于小麥自然群體全基因組關聯分析的赤霉病抗性關聯區間meta分析方法在小麥抗赤霉病育種中的應用:將獲得的meta分析結果用于小麥抗赤霉病育種中的分子標記輔助選擇。
15、本發明的有益效果如下:
16、(1)本發明提供的基于小麥自然群體全基因組關聯分析的赤霉病抗性關聯區間meta分析方法,與cn113113086a相比,創新在于利用已發表文獻的小麥自然群體,涉及2672個小麥品種,而且來源廣泛,通過對這些多樣化遺傳背景中的赤霉病抗性位點進行整合和meta分析,提高了位點關聯位點的可靠性,為培育抗赤霉病的小麥品種提供更多可用的可靠靶向位點。
17、(2)本發明提供的基于小麥自然群體全基因組關聯分析的赤霉病抗性關聯區間meta分析方法,通過收集和整合11篇關于小麥赤霉病自然群體全基因組關聯分析的文獻,將這些研究中定位到的關聯區間進行meta分析,得到64個抗赤霉病相關的meta區間。與現有技術中兩個親本衍生的小麥遺傳群體的meta研究的結果相比,4.7%是新的qtl位點,其余45.3%與小麥雙親本遺傳群體的研究結果一致。本發明的meta分析方法可以為抗赤霉病小麥品種選育提供更多精確的靶點及其關聯的標記,預期可進一步提高抗赤霉病分子標記輔助選擇育種效率。并且本發明的meta分析方法可擴展應用于不同作物重要性狀的gwas分析結果,具有廣泛的應用前景。
技術特征:
1.一種基于小麥自然群體全基因組關聯分析的赤霉病抗性關聯區間meta分析方法,其特征在于,包括以下步驟:
2.根據權利要求1所述的一種基于小麥自然群體全基因組關聯分析的赤霉病抗性關聯區間meta分析方法,其特征在于,步驟1)所述文獻篩選的標準為:①小麥群體只選用自然群體;②小麥赤霉病抗性性狀只關注小麥抗擴展以及抗毒素積累;③在gwas文章中只關注snp的結果;④選用重現性snp關聯結果。
3.根據權利要求2所述的一種基于小麥自然群體全基因組關聯分析的赤霉病抗性關聯區間meta分析方法,其特征在于,步驟1)所述文獻為11篇國內外公開發表的關于小麥赤霉病自然群體gwas研究的文獻,不包括連鎖分析和qtl驗證的文獻。
4.根據權利要求1所述的一種基于小麥自然群體全基因組關聯分析的赤霉病抗性關聯區間meta分析方法,其特征在于,步驟4)所述篩選的標準為:①qtl置信度的積累曲線聚類成峰,模型可以擬合;②一個ammqtl至少有2個完全或部分重疊的原始物理區間。
5.根據權利要求4所述的一種基于小麥自然群體全基因組關聯分析的赤霉病抗性關聯區間meta分析方法,其特征在于,步驟4)所述meta分析結果包括64個meta關聯區間,其中35個是與小麥抗赤霉病關聯的新的穩定meta關聯區間,其余29個與小麥雙親本衍生的遺傳群體meta研究結果一致。
6.根據權利要求5所述的一種基于小麥自然群體全基因組關聯分析的赤霉病抗性關聯區間meta分析方法,其特征在于,所述35個與小麥抗赤霉病關聯的新的穩定meta關聯區間的信息如下:
7.如權利要求1-6任一項所述的一種基于小麥自然群體全基因組關聯分析的赤霉病抗性關聯區間meta分析方法在小麥抗赤霉病育種中的應用,其特征在于,
技術總結
本發明公開了一種基于小麥自然群體全基因組關聯分析的赤霉病抗性關聯區間Meta分析方法和應用,通過Meta分析整合多篇赤霉病抗性全基因組關聯分析的結果,得到64個抗赤霉病相關的Meta區間。與現有技術中雙親本衍生的小麥遺傳群體的Meta研究的結果相比,54.7%是新的QTL位點,其余45.3%與小麥雙親本遺傳群體的研究結果一致。本發明的Meta分析方法可以為抗赤霉病小麥品種選育提供更多精確的靶點及其關聯的標記,預期可進一步提高抗赤霉病分子標記輔助選擇育種效率。并且該Meta分析方法可擴展應用于不同作物重要性狀的GWAS分析結果,具有廣泛的應用前景。
技術研發人員:李韜,王瀟,薛文婷,李磊,孫政璽,黃盼盼
受保護的技術使用者:揚州大學
技術研發日:
技術公布日:2025/4/24
- 還沒有人留言評論。精彩留言會獲得點贊!