一種2,4,6?三苯基嘧啶類衍生物的合成方法與流程
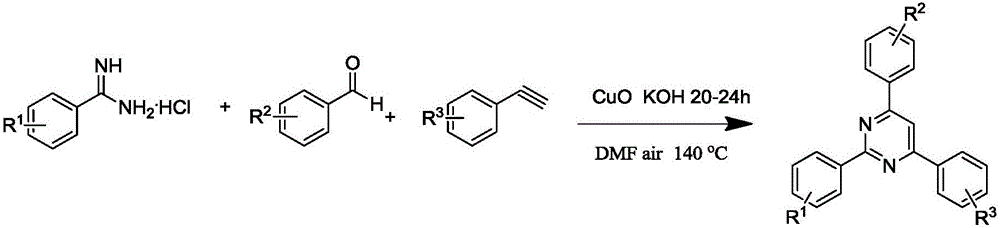
本發明屬于有機合成方法領域,具體涉及一種2,4,6-三苯基嘧啶類衍生物的合成方法。
技術背景
嘧啶類化合物是指分子結構中含有兩個氮原子的六元雜環化合物,與噠嗪、吡嗪互為同分異構體。嘧啶類化合物因具有不同的生物活性,如殺蟲、殺菌、除草、抗病毒等。從而引起人們對這類化合物的廣泛興趣并進行了深入研究。例如維生素B、磺胺嘧啶、磺胺異二甲嘧啶、磺胺間二甲氧嘧啶;農藥原料,如二嗪農、嘧啶醇、滅瘟素、除草定、環菌靈、甲菌定、乙菌定、磺嘧菌靈、比嗪農、抗蚜威、乙基蟲螨磷、蟲螨磷、多氧霉素、嘧啶威、嘧啶磷、氧嘧啶磷、嘧菌醇、異嘧菌醇、尿嘧啶等由于嘧啶化合物具有高效、對人類低毒等優點,因此其分子設計、合成與生物活性研究仍然是當今綠色農藥創制的一個熱點。
2,4,6-三苯基嘧啶是嘧啶環上的三個氫原子被其他原子或基團所取代,由于結構特點,在生物活性和藥理性質方面有著廣泛的應用,因此探索發展2,4,6-三苯基嘧啶的合成方法與技術任然是有機化學領域重要的研究課題之一。
目前文獻中介紹的合成2,4,6-三苯基嘧啶的方法主要有以下幾種:
(一)使用復雜的催化劑A催化芐脒鹽酸鹽和兩組分的醇一鍋法反應合成2,4,6-三苯基嘧啶衍生物。
該反應做到了三組分一鍋法反應,且運用兩種醇反應,符合綠色化學理念,但催化劑合成復雜,制約了其工業發展。
(二)使用五氯化鈮(NbCl5)催化腈和炔反應合成2,4,6-三苯基嘧啶衍生物。
此法反應條件溫和,但兩組分腈制約嘧啶的取代基相同,且產率較低。
(三)2012年Jean-Marc Campagne課題組利用β-enaminones作為中間體和酰胺進行反應得到2,4,6-三取代嘧啶衍生物。
該合成方法需要對中間體進行保護基團保護,且反應分步進行,合成效率不高。
(四)以芐脒鹽酸鹽和丙炔醇類化合物在微波條件反應合成2,4,6-三取代嘧啶衍生物。
這種方法利用微波條件,條件簡單但不利于工業擴大生產且反應過程使用MnO2重金屬,污染環境。
(五)使用醋酸鈀和碘化亞銅催化芐脒鹽酸鹽、碘苯、炔合成路線合成目標產物
但該合成方法使用了貴金屬Pd做催化劑,三叔丁基磷做配體,且反應產率一般。
綜上所述,現代合成2,4,6-三取代嘧啶化合物的方法很多,但有些合成方法4,6位取代基相同,不能做到不同取代基產物;有些使用貴金屬鈀作為催化劑,成本較高;有些合成步驟較為復雜或產率較低;因此提供一種合成高效,底物適用性廣的新型合成方法十分必要。
技術實現要素:
針對現有技術上的不足,本發明旨在提供一種2,4,6-三苯基嘧啶類衍生物的合成方法,在空氣條件下利用氧化銅催化芐脒鹽酸鹽、醛、炔三組分一鍋法反應,合成方法成本低,原料易得,底物普適性高。
本發明提供的一種2,4,6-三苯基嘧啶類衍生物的合成方法,包括以下步驟:
A、在空氣條件下,混合芐脒鹽酸鹽、醛和炔,然后加入催化劑、堿和溶劑,加熱反應;
B、反應結束后,將產物分離、干燥、純化后得到2,4,6-三苯基嘧啶類衍生物。
步驟A中所述芐脒鹽酸鹽、醛、炔、催化劑、堿的物質的量比為1:1.1-1.3:1.2-1.5:0.1:2-3。
步驟A中所述芐脒鹽酸鹽結構為:
式中R1為4-H、4-CH3、4-Cl、4-Br或4-CF3中的一種。
步驟A中所述醛結構為:
式中R2選自4-H、4-CH3、4-OCH3、4-Cl、3-Cl、2-Cl、4-Br、4-F或4-CF3中的一種。
步驟A中所述炔結構為:
式中R3選自4-H、4-CH3、4-OCH3、4-Cl、4-Br或4-F中的一種。
步驟A中所述加熱反應是指:在120℃-140℃條件下反應20-24小時;
步驟A中所述催化劑為氧化銅;所述堿為氫氧化鉀、氫氧化鈉、碳酸銫或碳酸鉀中的一種;所述溶劑為N,N-二甲基甲酰胺、二甲基亞砜或N,N-二甲基乙酰胺。芐脒鹽酸鹽在溶劑中的濃度為0.1-0.5mol/L。
步驟B具體為:將所得產物用乙酸乙酯萃取,然后無水硫酸鎂干燥,減壓濃縮后,得粗產物,以體積比純石油醚:乙酸乙酯=50:1為展開劑,通過柱層析分離得到2,4,6-三苯基嘧啶類衍生物。
步驟B所得2,4,6-三苯基嘧啶類衍生物結構式為式中,R1R1為4-H、4-CH3、4-Cl、4-Br或4-CF3;R2選自4-H、4-CH3、4-OCH3、4-Cl、3-Cl、2-Cl、4-Br、4-F或4-CF3;R3選自4-H、4-CH3、4-OCH3、4-Cl、4-Br或4-F。
本發明中,首先醛與離去鹽酸的脒進行了縮合反應生成亞胺,緊接著炔通過質子的轉移與亞胺結合形成嘧啶中間體,最后在氧氣的作用下脫H質子得到六元環2,4,6-嘧啶。本發明控制原料用量比,保證產率,以芐脒鹽酸鹽為反應基準,所以醛和炔是過量的,首先芐脒鹽酸鹽和醛進行縮合,所以1.2-1.3equiv的量可以使芐脒鹽酸鹽較為充分的轉化,炔的量加到1.3-1.5equiv,是因為炔易揮發,也是確保最大程度的進行反應,保證最大化的產物收率。
與現有技術相比,本發明具有以下優點:(1)沒有使用貴金屬,有機氧化劑,而是利用商品化氧化銅作催化劑,節約成本;(2)利用芐脒鹽酸鹽、醛、炔為反應原料,原料易得;(3)該合成方法效率高,使用范圍廣,底物適用性廣。
附圖說明
圖1為反應方程式;
式中,R1為4-H、4-CH3、4-Cl、3-Br或4-CF3;R2選自4-H、4-CH3、4-OCH3、4-Cl、3-Cl、2-Cl、4-Br、4-F或4-CF3;R3選自4-H、4-CH3、4-OCH3、4-Cl、4-Br、或4-F;
圖2為實施例1核磁共振氫譜;
圖3為實施例1核磁共振碳譜;
圖4為實施例2核磁共振氫譜;
圖5為實施例2核磁共振碳譜;
圖6為實施例3核磁共振氫譜;
圖7為實施例3核磁共振碳譜;
圖8為實施例4核磁共振氫譜;
圖9為實施例4核磁共振碳譜;
圖10為實施例5核磁共振氫譜;
圖11為實施例5核磁共振碳譜;
圖12為實施例6核磁共振氫譜;
圖13為實施例6核磁共振碳譜;
圖14為實施例7核磁共振氫譜;
圖15為實施例7核磁共振碳譜;
圖16為實施例8核磁共振氫譜;
圖17為實施例9核磁共振氫譜;
圖18為實施例9核磁共振碳譜。
具體實施方式
實施例1
2,4,6-三苯基嘧啶的合成,包括以下步驟:
A、取0.3mmol芐脒鹽酸鹽和0.39mmol苯甲醛、0.45mmol苯乙炔混合,再往其加入0.03mmol CuO,0.9mmol KOH和2-3ml DMF,在140℃下攪拌反應24小時;
B、所得產物用乙酸乙酯萃取,然后無水硫酸鎂干燥,減壓濃縮后,得粗產物,用硅膠柱層析(溶劑體積比為石油醚:乙酸乙酯=50:1)純化得白色固體即2,4,6-三苯基嘧啶,產率85%,熔點為182℃。
核磁表征數據:
1H NMR(300MHz,CDCl3):δ7.48-7.50(m,9H);7.97(s,1H),8.24–8.25(m,4H),8.67–8.69(m,2H);
13C NMR(75MHz,CDCl3):δ110.35,127.33,128.47,128.51,128.65,128.94,130.68,130.82,137.51,138.10,164.50,164.78。
實施例2
4-(4-氯苯基)-2,6-二苯基嘧啶的合成,包括以下步驟:
A、0.3mmol對芐脒鹽酸鹽和0.36mmol對氯苯甲醛、0.45mmol苯乙炔,再往其加入0.03mmol CuO,0.9mmol KOH以及2-3ml DMF,在140℃下攪拌反應24小時后,得產物;
B、將產物用乙酸乙酯進行萃取,干燥得到粗產物,將粗產物用硅膠柱層析(溶劑體積比為石油醚:乙酸乙酯=50:1)純化得白色固體4-(4-氯苯基)-2,6-二苯基嘧啶。產率81%,165-167℃。
核磁表征數據:
1H NMR(300MHz,CDCl3):δ7.47-7.50(t,8H),7.79(s,1H),8.18-8.24(m,4H),8.66(t,J=3Hz,2H).
13C NMR(75MHz,CDCl3):δ109.91,127.29,128.49,128.95,129.13,135.89,136.97,137.31,137.95,163.42,164.51,164.89.
實施例3
2-(4-氯苯基)-4-苯基-6-(對甲苯基)嘧啶的合成,包括以下步驟:
A、取0.3mmol芐脒鹽酸鹽和0.39mmol對氯苯甲醛、0.40mmol對甲基苯乙炔,再往其加入0.03mmol CuO,0.9mmol KOH以及2-3ml DMF,在140℃下攪拌反應24小時;
B、將產物用乙酸乙酯進行萃取,干燥得到粗產物,將粗產物用硅膠柱層析(溶劑體積比為石油醚:乙酸乙酯=50:1)純化得白色固體即2-(4-氯苯基)-4-苯基-6-(對甲苯基)嘧啶,產率76%,熔點為164℃。
核磁表征數據:
1H NMR(300MHz,CDCl3):δ2.41(s,3H),7.30-7.33(d,J=9.0Hz,2H),7.48-7.49(d,J=3Hz,2H),7.90(s,1H),8.12-8.20(m,4H),8.64-8.65(d,J=3Hz,2H);
13C NMR(75MHz,CDCl3):δ109.63,127.20,128.48,128.57,129.13,129.69,130.71,134.51,136.04,136.89,138.04,141.34,163.35,164.48,164.88。
實施例4
4-(2-氯苯基)-2,6-二苯基嘧啶的合成,包括以下步驟:
A、取1.5mmol芐脒鹽酸鹽和1.9mmol鄰氯苯甲醛、2.25mmol苯乙炔,再往其加入0.15mmol CuO,4.5mmol KOH以及3-4ml DMF,在140℃下攪拌反應24小時得到產物;
B、將產物用乙酸乙酯進行萃取,干燥得到粗產物,將粗產物用硅膠柱層析(溶劑體積比為石油醚:乙酸乙酯=50:1)純化得白色固體即4-(2-氯苯基)-2,6-二苯基嘧啶。產率70%,熔點為124-125℃。
核磁表征數據:
1H NMR(300MHz,CDCl3):δ7.38-7.51(m,9H),7.79-7.81(t,1H),7.97(s,1H),8.23(d,J=3.9Hz,2H),8.62(d,J=3.6Hz,2H);
13C NMR(75MHz,CDCl3):δ115.19,127.26,127.40,128.08,128.50,128.97,130.52,130.73,130.93,131.72,132.45,137.26,137.56,137.93,163.90,164.69。
實施例5
4-(3-氯苯基)-2,6-二苯基嘧啶的合成,包括以下步驟:
A、取2mmol芐脒鹽酸鹽和2.6mmol間氯苯甲醛、3mmol苯乙炔,再往其加入0.2mmol CuO,6mmol KOH以及6-8ml DMF,在140℃下攪拌反應24小時后,得產物;
B、將產物用乙酸乙酯進行萃取,干燥得到粗產物,將粗產物用硅膠柱層析(溶劑體積比為石油醚:乙酸乙酯=50:1)純化得白色固體即4-(3-Cl苯基)-2,6-二苯基嘧啶,產率52%,熔點為147-148℃.
核磁表征數據:
1H NMR(300MHz,CDCl3):δ7.46-7.50(m,8H)7.94(s,1H),8.11(d,J=5.7Hz,1H,)8.24,(s,3H),8.67(d,J=4.8Hz,2H)
13C NMR(75MHz,CDCl3):δ110.218,125.34,127.31,127.42,128.51,128.66,128.96,130.16,130.71,130.83,130.98,135.08,137.23,137.87,139.33,163.24,164.57,164.98.
實施例6
4-(對甲苯基)-2,6-二苯基嘧啶的合成,包括以下步驟:
A、取3mmol芐脒鹽酸鹽和3.9mmol對甲基苯甲醛、4.5mmol苯乙炔,再往其加入0.3mmol CuO,9mmol KOH以及6-8ml DMF,在140℃下攪拌反應24小時,得到產物;
B、將產物用乙酸乙酯進行萃取,干燥得到粗產物,將粗產物用硅膠柱層析(溶劑體積比為石油醚:乙酸乙酯=50:1)純化得白色固體即4-(對甲苯基)-2,6-二苯基嘧啶,產率83.1%,熔點為152-154℃.
核磁表征數據:
1H NMR(300MHz,CDCl3):δ2.41(s,3H),7.32(d,J=7.5Hz),7.48-7.50(m,6H),7.50(s,1H),8.15(d,J=7.8Hz,2H),8.24(d,J=5Hz),8.67(d,J=6Hz).
13C NMR(75MHz,CDCl3):δ21.51,109.99,127.23,127.32,128.44,128.50,128.91,129.67,130.60,130.72,134.70,137.64,138.23,141.18,164.42,164.62,164.69.
實施例7
2,4-二苯基-6-(2-噻吩基)嘧啶的合成,包括以下步驟:
A、取0.15mmol芐脒鹽酸鹽和0.18mmol 2-噻吩甲醛、0.225mmol苯乙炔,再往其加入0.015mmol CuO,0.45mmol KOH以及1-2ml DMF,在120℃下攪拌反應24小時得到產物;
B、將產物用乙酸乙酯進行萃取,干燥得到粗產物,將粗產物用硅膠柱層析(溶劑體積比為石油醚:乙酸乙酯=50:1)純化得白色固體,2,4-二苯基-6-(2-噻吩基),嘧啶產率86.3%,熔點為169-172℃
1H NMR(300MHz,CDCl3):δ7.17(d,J=3.6Hz,2H),7.48-7.50(m,6H),7.81(s,1H),7.89(s,1H),8.21(d,J=3.9Hz,2H),8.63(d,J=4.5Hz 2H).
13C NMR(75MHz,CDCl3):δ108.44,127.09,127.26,128.33,128.47,128.63,128.92,129.82,130.76,130.85,137.34,137.77,143.39,159.70,164.45,164.56.
實施例8
4,6-二苯基-2-(對甲苯基)嘧啶的合成,包括以下步驟:
A、0.3mmol對甲基芐脒鹽酸鹽和0.39mmol苯甲醛、0.45mmol苯乙炔,再往其加入0.03mmol CuO,0.9mmol KOH以及2-3ml DMF,在140℃下攪拌反應24小時,得到產物;
B、將產物用乙酸乙酯進行萃取,干燥得到粗產物,將粗產物用硅膠柱層析(溶劑體積比為石油醚:乙酸乙酯=50:1)純化得白色固體即4,6-二苯基-2-(對甲苯基)嘧啶。產率為88%,熔點為182-185℃
1H NMR(300MHz,CDCl3):δ2.41(s,3H),7.30(d,J=7.8Hz),7.50-7.52(d,6H),7.94(s,1H),8.24(t,J=5.4Hz,4H),8.58(d,J=7.8Hz,2H);
13C NMR(75MHz,CDCl3):δ21.60,110.06,127.31,128.48,128.91,129.94,130.72,135.51,137.67,140.85,164.67.
實施例9
4,6-二苯基-2-(對三氟甲基)嘧啶的合成,包括以下步驟:
A、0.3mmol對三氟甲基芐脒鹽酸鹽和0.39mmol苯甲醛、0.45mmol苯乙炔,再往其加入0.03mmol CuO,0.9mmol KOH以及2-3ml DMF,在140℃下攪拌反應24小時,得到產物;
B、將產物用乙酸乙酯進行萃取,干燥得到粗產物,將粗產物用硅膠柱層析(溶劑體積比為石油醚:乙酸乙酯=50:1)純化得白色固體4,6-二苯基-2-(對三氟甲基)嘧啶。產率為84.5%,熔點197-199℃
1H NMR(500MHz,CDCl3):δ7.58-7.61(m,6H),7.79(d,2H,8.5Hz),8.08(s,1H),8.29(t,4H),8.84(d,2H,8Hz).
13C NMR(75MHz,CDCl3):δ110.94,125.34,125.39,127.29,128.74,129.01,131.03,132.37,137.16,141.41,163.17,164.95.
以上詳細描述了本發明的優選實施方式。但是,本發明并不限于上述實施方式中的具體細節。只要在本發明的技術構思范圍內,可以對本發明的技術方案進行多種簡單變型的均屬于本發明的保護范圍。
- 還沒有人留言評論。精彩留言會獲得點贊!