金屬框架印跡材料及其制備方法與流程
本發(fā)明涉及納米材料技術(shù)領(lǐng)域,且特別涉及一種金屬框架印跡材料及其制備方法。
背景技術(shù):
有機磷農(nóng)藥由于其殺蟲效率高、價格較低、易降解等特性,已成為我國目前使用量最大的農(nóng)藥之一。然而不合理、不規(guī)范使用有機磷農(nóng)藥導(dǎo)致有機磷在環(huán)境中殘留、部分農(nóng)產(chǎn)品中有機磷殘留超標(biāo),對環(huán)境和人類健康都造成了威脅。因此,開發(fā)快速、準(zhǔn)確、靈敏的有機磷農(nóng)藥殘留的檢測方法,對保障食品安全和人類健康具有重要意義。
目前,研究者們已經(jīng)開發(fā)了高效液相色譜(HPLC)法、氣相色譜(GC)法、薄層色譜(TLC)法、氣相色譜-質(zhì)譜聯(lián)用(GC-MS)法、液相色譜-質(zhì)譜聯(lián)用(LC-MS)法和毛細(xì)管電泳法(CE)等儀器方法用于有機磷的檢測,這些方法需用到固相萃取、固相微萃取、液液萃取等前處理技術(shù)。然而傳統(tǒng)的農(nóng)藥樣品前處理技術(shù)缺乏選擇性、其操作往往較為繁瑣,檢測靈敏度和準(zhǔn)確度易受基質(zhì)效應(yīng)干擾。
分子印跡聚合物(MIPs)是一類具有較強分子識別能力的新型高分子仿生材料,具有穩(wěn)定性好、吸附性能強、特異識別性、重復(fù)使用等特點,非常適于從復(fù)雜基質(zhì)中分離純化特定痕量目標(biāo)物。目前的研究工作中,大多聚合物往往只識別單一有機磷化合物,不能滿足有機磷農(nóng)藥往復(fù)配方向發(fā)展的殘留檢測趨勢。
技術(shù)實現(xiàn)要素:
本發(fā)明的目的在于提供一種金屬框架印跡材料,以磁性金屬有機骨架材料為載體,使得該金屬框架印跡材料具有更好的穩(wěn)定性、吸附性、催化性,識別性能好、選擇性高、吸附容量大,可同時對環(huán)境及食品中多種有機磷類農(nóng)藥選擇性去除和富集。
本發(fā)明的另一目的在于提供一種金屬框架印跡材料的制備方法,該方法制備工藝簡單、成本低廉、環(huán)境友好度高、更易規(guī)模化。
本發(fā)明解決其技術(shù)問題是采用以下技術(shù)方案來實現(xiàn)的。
本發(fā)明提出一種金屬框架印跡材料的制備方法,包括以下步驟:將由對苯二甲酸與磁性Fe3O4納米粒子原位反應(yīng)制備的磁性金屬有機骨架化合物分散于溶劑中,加入模板分子、功能單體,交聯(lián)反應(yīng)后洗去模板分子;模板分子包括二嗪農(nóng)、對硫磷中的一種或兩種;功能單體包括3-氨丙基三乙氧基硅烷和苯三甲氧基硅烷中的一種或兩種。
本發(fā)明提出一種金屬框架印跡材料,由上述方法制備而成。
本發(fā)明實施例的一種金屬框架印跡材料及其制備方法的有益效果是:
本發(fā)明以磁性金屬有機骨架材料為載體制備金屬框架印跡材料,不僅使得材料具有特異識別性、良好的分散性和穩(wěn)定性、較好的吸附能力,可實現(xiàn)對多種有機磷農(nóng)藥的同時分離富集。還可利用材料的超順磁性實現(xiàn)磁分散固相萃取技術(shù),大大簡化了樣品前處理過程,擴展了現(xiàn)有金屬有機骨架材料的應(yīng)用范圍。該制備方法工藝簡單、成本低廉、環(huán)境友好度高、更易規(guī)模化。
附圖說明
為了更清楚地說明本發(fā)明實施例的技術(shù)方案,下面將對實施例中所需要使用的附圖作簡單地介紹,應(yīng)當(dāng)理解,以下附圖僅示出了本發(fā)明的某些實施例,因此不應(yīng)被看作是對范圍的限定,對于本領(lǐng)域普通技術(shù)人員來講,在不付出創(chuàng)造性勞動的前提下,還可以根據(jù)這些附圖獲得其他相關(guān)的附圖。
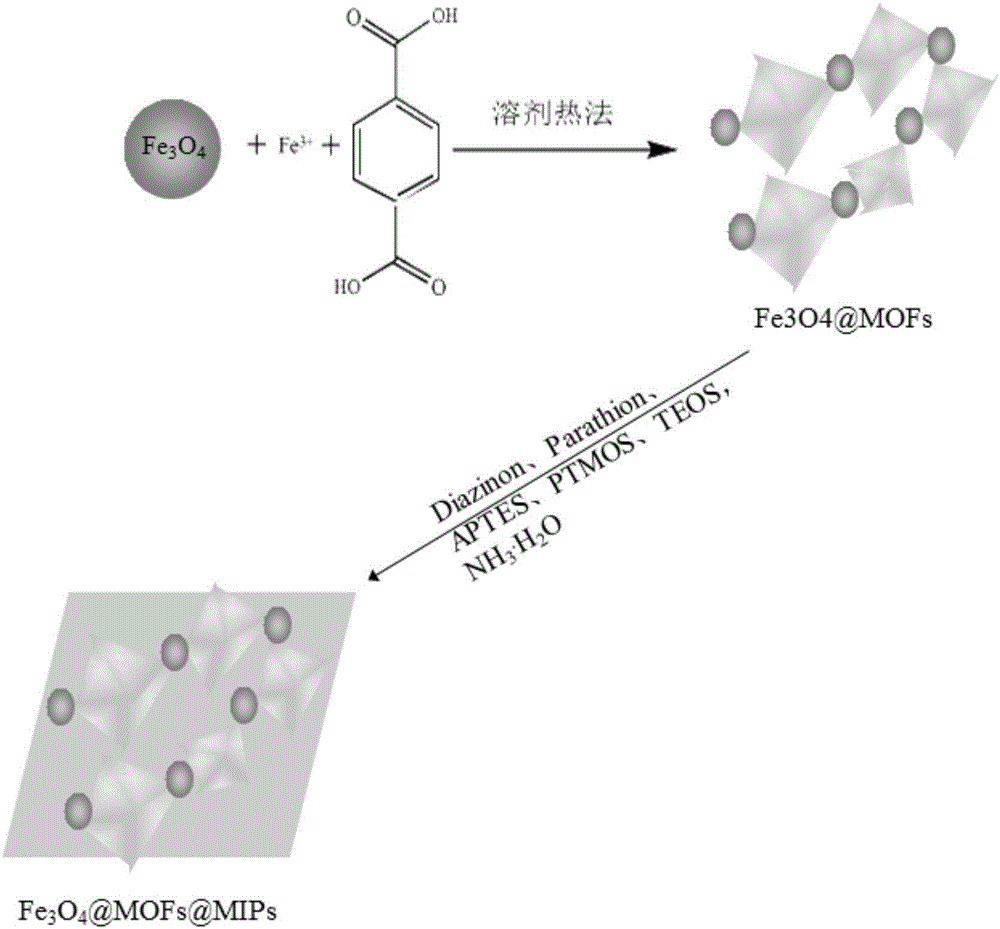
圖1為本發(fā)明實施例的金屬框架印跡材料的制備流程示意圖;
圖2為本發(fā)明實施例2的金屬框架印跡材料的掃描電子顯微鏡圖;
圖3為本發(fā)明實施例2的磁性Fe3O4納米粒子、磁性金屬有機骨架化合物、金屬框架印跡材料的磁滯回線;
圖4為本發(fā)明實施例2的金屬框架印跡材料和對比例1的金屬框架非印跡材料對有機磷農(nóng)藥二嗪農(nóng)的動態(tài)吸附曲線;
圖5為本發(fā)明實施例2的金屬框架印跡材料和對比例1的金屬框架非印跡材料對6種有機磷農(nóng)藥的吸附作用圖。
具體實施方式
為使本發(fā)明實施例的目的、技術(shù)方案和優(yōu)點更加清楚,下面將對本發(fā)明實施例中的技術(shù)方案進行清楚、完整地描述。實施例中未注明具體條件者,按照常規(guī)條件或制造商建議的條件進行。所用試劑或儀器未注明生產(chǎn)廠商者,均為可以通過市售購買獲得的常規(guī)產(chǎn)品。
下面對本發(fā)明實施例的一種金屬框架印跡材料及其制備方法進行具體說明。
本發(fā)明實施例提供的一種金屬框架印跡材料的制備方法,包括以下步驟:
制備磁性金屬有機骨架化合物;
將磁性Fe3O4納米粒子超聲分散到含有FeCl3的DMF溶液中,加入含有對苯二甲酸的DMF溶液,超聲分散均勻后,將混合溶液置于反應(yīng)釜的內(nèi)襯管中,110~150℃下反應(yīng)16~24h。待反應(yīng)結(jié)束后用無水乙醇洗滌反應(yīng)產(chǎn)物,去除反應(yīng)產(chǎn)物中的乙醇可溶物,55~65℃干燥10~14h,即得磁性金屬有機骨架化合物。因為FeCl3易吸水并且有強烈的吸水性,能吸收空氣里的水分而潮解,故FeCl3可選用FeCl3·6H2O。優(yōu)選地,磁性Fe3O4納米粒子、FeCl3、對苯二甲酸的質(zhì)量比為1~2:20~40:15~33。
其中,磁性Fe3O4納米粒子可購買,也可制備而得,本發(fā)明實施例中,具體的磁性Fe3O4納米粒子制備方法如下:將FeCl3·6H2O、FeCl2·4H2O和去離子水置于250mL三口燒瓶中,70~90℃氮氣保護下機械攪拌20~40min后,加入氨水。待反應(yīng)結(jié)束后冷卻至20~30℃,使用外加磁場收集磁性Fe3O4納米粒子,并用超純水和乙醇洗滌。優(yōu)選地,F(xiàn)eCl3·6H2O、FeCl2·4H2O、氨水的用量比為:4~8mmol:2~5mmol:5mL。
將磁性金屬有機骨架化合物超聲分散于溶劑中,溶劑包括乙腈、甲醇、氯仿中的至少一種。加入模板分子,模板分子包括二嗪農(nóng)、對硫磷中的一種或兩種;持續(xù)攪拌20~40min的同時加入功能單體,功能單體包括3-氨丙基三乙氧基硅烷和苯三甲氧基硅烷中的一種或兩種;再加入交聯(lián)劑四乙基硅烷和催化劑25%(V/W)氨水,30~50℃下反應(yīng)16~24h。持續(xù)攪拌20~40min是為了使磁性金屬有機骨架化合物、模板分子、功能單體充分混合,防止團聚或混合不均導(dǎo)致產(chǎn)物性質(zhì)不同。
功能單體的選擇主要取決于模板分子,功能單體需要能與模板分子通過非共價鍵相互作用,而且能與交聯(lián)劑反應(yīng)使模板分子鑲嵌其中并獲得預(yù)期的取向和定位。
交聯(lián)劑是固定模板分子和功能單體維持空穴的剛性程度的重要因素,也影響著傳質(zhì)作用。
溶劑既有溶解所有反應(yīng)試劑的作用,也是制備識別材料的致孔劑。溶劑的選擇和用量不僅影響功能單體和模板分子間的結(jié)合強度、動力學(xué)性質(zhì),也影響著對分子聚合物的形態(tài)和結(jié)構(gòu)。
磁性金屬有機骨架化合物、模板分子、功能單體的質(zhì)量比為50~100:76:0.03~0.16。磁性金屬有機骨架化合物、模板分子、功能單體、交聯(lián)劑、催化劑的用量比為:100~200mg:0.1~1mmol:0.05~0.7mL:0.45~2.25mL:0.3~0.8mL。
模板分子、功能單體和交聯(lián)劑用量的比例也對核殼型識別材料的性能有著重大影響。一般增大功能單體的比例可以充分的預(yù)組裝模板分子,但過高的功能單體比例會增加核殼型識別材料中非選擇性結(jié)合位點,以及可能由于功能單體的自我聚合而減少選擇性結(jié)合位點。交聯(lián)劑的比例較低時,核殼型識別材料會因為交聯(lián)度不夠而無法保持穩(wěn)定的空穴結(jié)構(gòu),使識別能力降低,而過高比例的交聯(lián)劑會因為核殼型識別材料中含有的功能單體數(shù)目低而減少選擇性結(jié)合位點,會破壞印跡效果。
反應(yīng)完后用磁鐵手機沉淀物,用體積比為1:8~10的乙酸、甲醇混合溶劑洗去模板分子,其中,可將乙酸、甲醇混合溶液與模板分子混合,攪拌后過濾,反復(fù)操作。真空干燥后得到金屬框架印跡材料。
該制備方法首先采用共沉淀法制備磁性四氧化三鐵納米粒子,然后采用原位法制備磁性金屬有機骨架化合物。進一步,以具有適當(dāng)功能基團的功能單體與模板分子通過非共價鍵結(jié)合,形成含有磁性金屬有機骨架化合物的單體模板分子復(fù)合物,交聯(lián)劑固定圍繞模板分子的功能單體,形成高度交聯(lián)的剛性聚合物,從而使功能單體上的功能官能團在空間結(jié)構(gòu)上固定下來,最后將模板分子脫去,得到與模板分子在官能團和空間結(jié)構(gòu)上互補的核殼型識別材料。
本發(fā)明還提供了一種核殼型識別材料,由上述方法制備而成。
以下結(jié)合實施例對本發(fā)明的特征和性能作進一步的詳細(xì)描述。
實施例1
請參考圖1,本實施例提供了一種金屬框架印跡材料,通過以下步驟制得:
將4mmolFeCl3、2mmolFeCl2和100mL去離子水置于250mL三口燒瓶中,70℃氮氣保護下機械攪拌20min后,加入5mL氨水。待反應(yīng)結(jié)束后冷卻至20℃,使用外加磁場收集磁性Fe3O4納米粒子,并用超純水和乙醇洗滌。
將50mg磁性Fe3O4納米粒子超聲分散到含有2.5g的FeCl3·6H2O的DMF溶液中,加入含有0.75g對苯二甲酸的DMF溶液,DMF溶液為50mL,超聲分散均勻后,將混合溶液置于反應(yīng)釜的內(nèi)襯管中,110℃下反應(yīng)16h。待反應(yīng)結(jié)束后用無水乙醇洗滌,55℃干燥10h,即得磁性金屬有機骨架化合物。
將100mg磁性金屬有機骨架化合物超聲分散于10mL乙腈中,加入0.1mmol模板分子二嗪農(nóng);持續(xù)攪拌20min的同時加入0.05mL功能單體3-氨丙基三乙氧基硅烷;再加入0.45mL交聯(lián)劑四乙基硅烷和0.3mL催化劑25%(V/W)氨水,30℃下反應(yīng)16h。反應(yīng)完后用磁鐵手機沉淀物,用體積比為1:8的乙酸、甲醇混合溶劑洗去模板分子,真空干燥后得到金屬框架印跡材料。
實施例2
請參考圖1,本實施例提供了一種金屬框架印跡材料,通過以下步驟制得:
將6mmolFeCl3·6H2O、2.5mmolFeCl2·4H2O和100mL去離子水置于250mL三口燒瓶中,80℃氮氣保護下機械攪拌30min后,加入5mL氨水。待反應(yīng)結(jié)束后冷卻至25℃,使用外加磁場收集磁性Fe3O4納米粒子,并用超純水和乙醇洗滌。
將75mg磁性Fe3O4納米粒子超聲分散到含有3.5g的FeCl3·6H2O的DMF溶液中,加入含有1.2g對苯二甲酸的DMF溶液,DMF溶液為100mL,超聲分散均勻后,將混合溶液置于反應(yīng)釜的內(nèi)襯管中,130℃下反應(yīng)20h。待反應(yīng)結(jié)束后用無水乙醇洗滌,60℃干燥12h,即得磁性金屬有機骨架化合物。
將150mg磁性金屬有機骨架化合物超聲分散于350mL乙腈、甲醇的混合溶液中,加入0.5mmol模板分子二嗪農(nóng)、對硫磷;持續(xù)攪拌30min的同時加入0.4mL功能單體3-氨丙基三乙氧基硅烷和苯三甲氧基硅烷;再加入1.35mL交聯(lián)劑四乙基硅烷和0.5mL催化劑25%(V/W)氨水,40℃下反應(yīng)20h。反應(yīng)完后用磁鐵手機沉淀物,用體積比為1:9的乙酸、甲醇混合溶劑洗去模板分子,真空干燥后得到金屬框架印跡材料。
實施例3
請參考圖1,本實施例提供了一種金屬框架印跡材料,通過以下步驟制得:
將8mmolFeCl3·6H2O、5mmolFeCl2·4H2O和100mL去離子水置于250mL三口燒瓶中,90℃氮氣保護下機械攪拌40min后,加入5mL氨水。待反應(yīng)結(jié)束后冷卻至30℃,使用外加磁場收集磁性Fe3O4納米粒子,并用超純水和乙醇洗滌。
將100mg磁性Fe3O4納米粒子超聲分散到含有5g的FeCl3·6H2O的DMF溶液中,加入含有1.65g對苯二甲酸的DMF溶液,DMF溶液為150mL,超聲分散均勻后,將混合溶液置于反應(yīng)釜的內(nèi)襯管中,150℃下反應(yīng)24h。待反應(yīng)結(jié)束后用無水乙醇洗滌,65℃干燥14h,即得磁性金屬有機骨架化合物。
將200mg磁性金屬有機骨架化合物超聲分散于600mL乙腈、甲醇、氯仿的混合溶液中,加入1mmol模板分子對硫磷;持續(xù)攪拌40min的同時加入0.7mL功能單體苯三甲氧基硅烷;再加入2.25mL交聯(lián)劑四乙基硅烷和0.8mL催化劑25%(V/W)氨水,50℃下反應(yīng)24h。反應(yīng)完后用磁鐵手機沉淀物,用體積比為1:10的乙酸、甲醇混合溶劑洗去模板分子,真空干燥后得到金屬框架印跡材料。
實施例4
請參考圖1,本實施例提供了一種金屬框架印跡材料,通過以下步驟制得:
將6mmolFeCl3·6H2O、3mmolFeCl2·4H2O和100mL去離子水置于250mL三口燒瓶中,80℃氮氣保護下機械攪拌30min后,加入5mL氨水。待反應(yīng)結(jié)束后冷卻至25℃,使用外加磁場收集磁性Fe3O4納米粒子,并用超純水和乙醇洗滌。
將75mg磁性Fe3O4納米粒子超聲分散到含有5g的FeCl3·6H2O的DMF溶液中,加入含有1.5g對苯二甲酸的DMF溶液,DMF溶液為150mL,超聲分散均勻后,將混合溶液置于反應(yīng)釜的內(nèi)襯管中,120℃下反應(yīng)20h。待反應(yīng)結(jié)束后用無水乙醇洗滌,60℃干燥12h,即得磁性金屬有機骨架化合物。
將100mg磁性金屬有機骨架化合物超聲分散于100mL乙腈、氯仿的混合溶液中,加入0.5mmol模板分子二嗪農(nóng)、對硫磷;持續(xù)攪拌30min的同時加入0.7mL功能單體3-氨丙基三乙氧基硅烷和苯三甲氧基硅烷;再加入1.5mL交聯(lián)劑四乙基硅烷和0.3mL催化劑25%(V/W)氨水,30℃下反應(yīng)20h。反應(yīng)完后用磁鐵手機沉淀物,用體積比為1:10的乙酸、甲醇混合溶劑洗去模板分子,真空干燥后得到金屬框架印跡材料。
實施例5
請參考圖1,本實施例提供了一種金屬框架印跡材料,通過以下步驟制得:
將4mmolFeCl3·6H2O、2mmolFeCl2·4H2O和100mL去離子水置于250mL三口燒瓶中,70℃氦氣保護下機械攪拌40min后,加入5mL氨水。待反應(yīng)結(jié)束后冷卻至30℃,使用外加磁場收集磁性Fe3O4納米粒子,并用超純水和乙醇洗滌。
將100mg磁性Fe3O4納米粒子超聲分散到含有2.5g的FeCl3·6H2O的DMF溶液中,加入含有0.75g對苯二甲酸的DMF溶液,DMF溶液為75mL,超聲分散均勻后,將混合溶液置于反應(yīng)釜的內(nèi)襯管中,130℃下反應(yīng)20h。待反應(yīng)結(jié)束后用無水乙醇洗滌,65℃干燥14h,即得磁性金屬有機骨架化合物。
將100磁性金屬有機骨架化合物超聲分散于600mL甲醇、氯仿的混合溶液中,加入1mmol模板分子二嗪農(nóng)、對硫磷;持續(xù)攪拌20min的同時加入0.7mL功能單體3-氨丙基三乙氧基硅烷和苯三甲氧基硅烷;再加入0.45mL交聯(lián)劑四乙基硅烷和0.8mL催化劑25%(V/W)氨水,30℃下反應(yīng)16h。反應(yīng)完后用磁鐵手機沉淀物,用體積比為1:8的乙酸、甲醇混合溶劑洗去模板分子,真空干燥后得到金屬框架印跡材料。
對比例1
一種金屬框架非印跡材料,通過以下步驟制得:
將6mmolFeCl3·6H2O、2.5mmolFeCl2·4H2O和100mL去離子水置于250mL三口燒瓶中,80℃氮氣保護下機械攪拌30min后,加入5mL氨水。待反應(yīng)結(jié)束后冷卻至25℃,使用外加磁場收集磁性Fe3O4納米粒子,并用超純水和乙醇洗滌。
將75mg磁性Fe3O4納米粒子超聲分散到含有3.5g的FeCl3·6H2O的DMF溶液中,加入含有1.2g對苯二甲酸的DMF溶液,DMF溶液為100mL,超聲分散均勻后,將混合溶液置于反應(yīng)釜的內(nèi)襯管中,130℃下反應(yīng)20h。待反應(yīng)結(jié)束后用無水乙醇洗滌,60℃干燥12h,即得磁性金屬有機骨架化合物。
將150mg磁性金屬有機骨架化合物超聲分散于乙腈、甲醇、氯仿的混合溶液中,加入0.5mmol模板分子二嗪農(nóng)、對硫磷;持續(xù)攪拌30min的同時加入0.4mL功能單體3-氨丙基三乙氧基硅烷和苯三甲氧基硅烷;再加入1.35mL交聯(lián)劑四乙基硅烷和0.5mL催化劑25%(V/W)氨水,40℃下反應(yīng)20h。反應(yīng)完后用磁鐵手機沉淀物,用體積比為1:9的乙酸、甲醇混合溶劑洗去模板分子,真空干燥后得到金屬框架印跡材料。
試驗例1
選取實施例1~5的金屬框架印跡材料進行性能檢測,方法如下:
分別將10g實施例1~5的金屬框架印跡材料加入到5mL二嗪農(nóng)濃度為40μg/L的甲醇溶液中,室溫下震蕩20min后,通過外加磁場將上清液分離出來,經(jīng)有機濾膜處理后,用HPLC-MS/MS分離測定。測試結(jié)果如下:
表1 金屬框架印跡材料對二嗪農(nóng)的吸附量
由表可知,實施例1~5的金屬框架印跡材料的吸附量都較高,其中,實施例2的金屬框架印跡材料的吸附量最大,說明該金屬框架印跡材料具有更好的性能。
試驗例2
對實施例2的金屬框架印跡材料進行掃描電鏡檢測。請參照圖2,由圖可知,經(jīng)過表面修飾后制備得到的金屬框架印跡材料,在表面包覆一層分子印跡殼層。
試驗例3
分別對本發(fā)明實施例2的磁性Fe3O4納米粒子、磁性金屬有機骨架化合物、金屬框架印跡材料進行磁滯回線測試,結(jié)果見圖3。圖3中,以粗實線(a)表示磁性Fe3O4納米粒子的磁滯回線,以細(xì)點畫線(b)表示磁性金屬有機骨架化合物的磁滯回線,以粗點畫線(c)表示金屬框架印跡材料的磁滯回線。由圖3可知,實施例2中制備得到的金屬框架印跡材料的磁滯作用比較微弱,說明了包覆識別材料的磁性納米微球在室溫下具有超順磁性,移去外加磁場之后不會保留剩磁,從而使其能再次快速分散而不會發(fā)生團聚。
試驗例4
選取試驗例2、對比例1的金屬框架印跡材料進行性能檢測,方法如下:
(1)分別稱取二嗪農(nóng)diazinon、毒死蜱Chlorpyrifos、對硫磷Parathion、甲基對硫磷Parathion-methyl、殺螟硫磷fenitrothion、倍硫磷fenthion等環(huán)境內(nèi)分泌干擾物標(biāo)準(zhǔn)品10.0mg于6個10mL容量瓶中,用少量甲醇溶解后,用甲醇定容至刻度,制成1.0g/L的標(biāo)準(zhǔn)儲備液,4℃冰箱中保存?zhèn)溆谩?/p>
(2)將10mg金屬框架印跡材料和金屬框架非印跡材料分別加入到5mL二嗪農(nóng)濃度均為40μg/L的甲醇溶液中,室溫下震蕩30min后,通過外加磁場將上清液分離出來,經(jīng)有機濾膜處理后用HPLC-MS/MS分離測定。
(3)將10mg金屬框架印跡材料和金屬框架非印跡材料分別加入到5mL六種有機磷類農(nóng)藥濃度均為40μg/L的甲醇混合液中,室溫下震蕩8min后,通過外加磁場將上清液分離出來,經(jīng)有機濾膜處理后,用HPLC-MS/MS分離測定。
請參照圖4,圖4為金屬框架印跡材料和金屬框架非印跡材料對有機磷農(nóng)藥甲基對硫磷的動態(tài)吸附曲線,吸附量隨時間的增加而增加,達到一定時間后,吸附量變化較小,趨于穩(wěn)定。金屬框架印跡材料MIP的吸附量明顯大于金屬框架非印跡材料NIP,吸附速率較大,可以對有機磷農(nóng)藥二嗪農(nóng)快速提取。
請參照圖5,圖5為金屬框架印跡材料和金屬框架非印跡材料對6種有機磷農(nóng)藥的吸附作用圖,由圖5可知,金屬框架印跡材料MIP對6種有機磷農(nóng)藥的吸附量明顯大于金屬框架非印跡材料NIP,且對每種有機磷農(nóng)藥的吸附量都較高,具有較好的識別性。
以上所描述的實施例是本發(fā)明一部分實施例,而不是全部的實施例。本發(fā)明的實施例的詳細(xì)描述并非旨在限制要求保護的本發(fā)明的范圍,而是僅僅表示本發(fā)明的選定實施例。基于本發(fā)明中的實施例,本領(lǐng)域普通技術(shù)人員在沒有作出創(chuàng)造性勞動前提下所獲得的所有其他實施例,都屬于本發(fā)明保護的范圍。
- 還沒有人留言評論。精彩留言會獲得點贊!
- 具有磁性和葉酸雙重靶向的復(fù)合納米材料的制備方法與流程
- 基于納米金增敏的戊唑醇分子印跡膜電極及其電化學(xué)傳感器的制備、應(yīng)用和測試方法與流程
- 一種雙親性乙草胺磁性分子印跡聚合物及其制備方法和應(yīng)用與流程
- 一種磁性多孔分子印跡聚合物的制備方法與流程
- 超分子穴番?A負(fù)載鈀納米材料的制備方法及應(yīng)用與流程
- 一種三元磁性復(fù)合光催化納米材料及其制備方法和用途與流程
- 利用懸掛雙鍵聚合法制備磁性鄰苯二甲酸酯類印跡聚合物的制作方法與工藝
- 一種磁性納米吸油材料、其制備方法及用途與流程
- 磁性Fe3O4@PPy復(fù)合納米材料的制備方法與流程
- 一種分離粘稠血分子的磁性保健品的制作方法與工藝