一種硅基碳硼烷聚合物及其制備方法與流程
本發明涉及一種硅基碳硼烷聚合物及其制備方法,屬于精細化學品和耐高溫聚合物合成技術領域。
技術背景
sibcn陶瓷作為一種新型高性能陶瓷材料,比sic、si3n4、bn等二元體系陶瓷和sicn、sico等三元體系的陶瓷具有更優異的高溫穩定性和抗氧化性,有些sibcn陶瓷的高溫穩定性甚至可達到2000℃。正因如此,sibcn陶瓷數十年以來作為陶瓷涂層、陶瓷纖維、陶瓷基復合材料的研究方興未艾。此外還可用作陶瓷的粘接劑、電腦芯片的多層連接等,因此被廣泛應用于信息、電子、航空航天和軍事等領域。
sibcn陶瓷主要采用有機無機雜化聚合物聚硼硅氮烷(pbsz)經高溫裂解獲得。其中,聚硼硅氮烷的合成主要采用兩類合成路徑,分別為單體途徑和聚合物途徑。
單體途徑的特征是先利用含有硼或者硅的單元反應生成分別含有硼、硅的單體,然后再聚合成sibcn陶瓷前驅體,但副產物難去除,無法工業化。聚合物路徑的特征是利用含硅聚合物與含硼化合物反應,制得陶瓷前驅體,但聚合物途徑在前驅體制備收率及后續陶瓷化產率方面均明顯低于單體途徑,因此成本較高;或者先將多乙烯基環硅氮烷硼烷化,利用環硅氮烷的反應性交聯獲得陶瓷體,然而,將硅氮烷進行硼烷化的方法,由于使用的有機硼烷具有劇毒,生產受到限制,可操作性差,難以工業化實施。
技術實現要素:
本發明針對現有的聚硼硅氮烷陶瓷前驅體制備工藝或復雜或采用有毒原料,以及合成成本高等不足,提供了一種新型的新型硅基碳硼烷聚合物及其制備方法。所公開的嵌段聚合物由無毒的碳硼烷合成制得,由于特有的合成路線得到的網狀結構,擁有更高的陶瓷化產率,并且可由市售材料在非常溫和的條件下制得,適合放大生產,便于在工業中推廣應用。
本發明公開了一種硅基碳硼烷聚合物,其化學結構式如下:
其中,r=-ch3或-ph;m、n分別為3~50的整數。
本發明還公開了上述的硅基碳硼烷聚合物的制備方法,包括如下步驟:
(1)以1,1′-烴基硅甲撐-2,2′-雙(二甲基硅基)雙碳硼烷為原料,在堿性條件下、金屬催化劑下,進行反應制備1,1′-烴基硅甲撐-2,2′-雙(二甲基羥基硅基)雙碳硼烷;
(2)以二甲基氯硅烷、三乙烯基三甲基環三硅氮烷為原料,在金屬催化劑下,反應制備1,3,5-三甲基-1,3,5-三[β-(二甲基氯硅基)乙基]環三硅氮烷;
(3)以1,1′-烴基硅甲撐-2,2′-雙(二甲基羥基硅基)雙碳硼烷、1,3,5-三甲基-1,3,5-三[β-(二甲基氯硅基)乙基]環三硅氮烷為原料,在三乙胺存在下,制備所述硅基碳硼烷聚合物。
上述技術方案中,所述1,1′-烴基硅甲撐-2,2′-雙(二甲基硅基)雙碳硼烷為1,1′-甲基苯基硅甲撐-2,2′-雙(二甲基硅基)雙碳硼烷或1,1′-雙苯基硅甲撐-2,2′-雙(二甲基硅基)雙碳硼烷;所述金屬催化劑為鈀碳催化劑或鉑碳催化劑;所述堿性條件為氫氧化鈉、一水磷酸二氫鈉條件。
上述技術方案中,步驟(1)反應完成后,過濾反應液,然后濾液經萃取、水洗、干燥、除溶劑得到1,1′-烴基硅甲撐-2,2′-雙(二甲基羥基硅基)雙碳硼烷;步驟(3)反應完成后,過濾反應液,然后濾液經萃取、水洗、干燥、除溶劑得到所述硅基碳硼烷聚合物。
上述技術方案中,
步驟(1)為,將氫氧化鈉、一水磷酸二氫鈉與水混合制備緩沖液,再加入金屬催化劑、1,1′-烴基硅甲撐-2,2′-雙(二甲基硅基)雙碳硼烷,反應制備1,1′-烴基硅甲撐-2,2′-雙(二甲基羥基硅基)雙碳硼烷;
步驟(2)為,惰性氣氛下,在三乙烯基三甲基環三硅氮烷溶液中加入金屬催化劑;然后加入二甲基氯硅烷溶液;反應制備1,3,5-三甲基-1,3,5-三[β-(二甲基氯硅基)乙基]環三硅氮烷;
步驟(3)為,將含有三乙胺的1,1′-烴基硅甲撐-2,2′-雙(二甲基羥基硅基)雙碳硼烷滴加入1,3,5-三甲基-1,3,5-三[β-(二甲基氯硅基)乙基]環三硅氮烷中,反應制備所述硅基碳硼烷聚合物。
上述技術方案中,
步驟(1)為,將氫氧化鈉與水混合,再加入一水磷酸二氫鈉混合制備緩沖液,再加入醚類溶劑以及金屬催化劑;然后加入1,1′-烴基硅甲撐-2,2′-雙(二甲基硅基)雙碳硼烷的芳烴溶液,反應制備1,1′-烴基硅甲撐-2,2′-雙(二甲基羥基硅基)雙碳硼烷;
步驟(2)為,惰性氣氛下,在三乙烯基三甲基環三硅氮烷芳烴溶液中加入金屬催化劑;然后加入二甲基氯硅烷芳烴溶液;反應制備1,3,5-三甲基-1,3,5-三[β-(二甲基氯硅基)乙基]環三硅氮烷;
步驟(3)為,將含有三乙胺的1,1′-烴基硅甲撐-2,2′-雙(二甲基羥基硅基)雙碳硼烷芳烴溶液滴加入1,3,5-三甲基-1,3,5-三[β-(二甲基氯硅基)乙基]環三硅氮烷中,反應制備所述硅基碳硼烷聚合物。
上述技術方案中,所述醚類溶劑為1,4-二氧六環,所述芳烴為甲苯和/或二甲苯;所述惰性氣氛為氮氣氣氛。
上述技術方案中,
步驟(1)中,氫氧化鈉、水、一水磷酸二氫鈉、金屬催化劑、1,1′-烴基硅甲撐-2,2′-雙(二甲基硅基)雙碳硼烷的質量比為(0.2~2)∶(20~200)∶(0.2~5)∶(0.1~2)∶(0.5~5);
步驟(2)中,三乙烯基三甲基環三硅氮烷、金屬催化劑、二甲基氯硅烷的質量比為(0.5~5)∶(0.001~0.2)∶(0.5~5);
步驟(3)中,三乙胺、1,1′-烴基硅甲撐-2,2′-雙(二甲基羥基硅基)雙碳硼烷的質量比為(0.5~10)∶(1~5)。
上述技術方案中,
步驟(1)中,反應溫度為25℃~65℃,反應時間為1~48小時;
步驟(2)中,反應溫度為25℃~85℃,反應時間為1~48小時;
步驟(3)中,反應溫度為25℃~75℃,反應時間為1~48小時。
本發明還公開了1,1′-烴基硅甲撐-2,2′-雙(二甲基硅基)雙碳硼烷的制備,惰性氣體下,向烴基二氯硅烷溶液內滴加乙炔基溴化鎂格氏試劑,于35~45℃下反應;然后滴加癸硼烷溶液,于80~90℃下反應,得到烴基硅甲撐雙碳硼烷;惰性氣體下,冰水浴條件下,向烴基硅甲撐雙碳硼烷溶液內滴加正丁基鋰溶液,冰水浴條件下反應;然后滴加二甲基氯硅烷溶液,于冰水浴條件下下反應,然后于室溫反應,得到1,1′-烴基硅甲撐-2,2′-雙(二甲基硅基)雙碳硼烷。烴基二氯硅烷、乙炔基溴化鎂格氏試劑、癸硼烷的質量比為(0.2~2)∶(0.5~5)∶(0.1~1);烴基硅甲撐雙碳硼烷、正丁基鋰、二甲基氯硅烷的質量比為(1~10)∶(0.2~2)∶(0.5~5)。
本發明還公開了一種sibcn陶瓷的制備方法,將上述的硅基碳硼烷聚合物經過成型處理得到sibcn陶瓷。通過現有方式實現硅基碳硼烷聚合物的成型處理,比如載體涂覆后風干,或者進一步熱處理等。
本發明還公開了一種sibcn陶瓷,由上述的硅基碳硼烷聚合物經過成型處理得到。
本發明具體反應式如下:
其中,r=-ch3或-ph;m、n分別為3~50的整數。
本發明公開的新型硅基碳硼烷聚合物,其以工業化原料三乙烯基三甲基環三硅氮烷為骨架支撐,通過與無毒的碳硼烷單體聚合反應引入耐熱單元,得到的預聚物以其四元體系和網狀交聯結構可以提供非常優異的耐熱性能和抗氧化性能,其簡易的合成路線以及溫和的反應條件,便于推廣應用于制備sibcn陶瓷材料。
制備硅基碳硼烷聚合物的具體操作步驟為:
(1)
a.將氫氧化鈉,去離子水和一水磷酸二氫鈉混合均勻,制備成緩沖溶液。
b.將緩沖溶液加入到反應器中,再加入1,4-二氧六環以及貴金屬催化劑,然后加入1,1′-烴基硅甲撐-2,2′-雙(二甲基硅基)雙碳硼烷溶于芳烴溶劑中所形成的溶液,進行羥基化反應。
c.反應結束后,過濾除去不溶的貴金屬催化劑,濾液加入萃取劑萃取。
d.合并萃取液,用去離子水洗至中性,經干燥、旋蒸,得到1,1′-烴基硅甲撐-2,2′-雙(二甲基羥基硅基)雙碳硼烷。
(2)
a.由二甲基氯硅烷溶解于除水芳烴溶劑中,配制二甲基氯硅烷溶液,備用。
b.在反應器中投入三乙烯基三甲基環三硅氮烷和除水芳烴溶劑,氮氣保護下,加入鉑催化劑和上述a中配制好的二甲基氯硅烷溶液,在氮氣保護下保溫反應一定時間,得到1,3,5-三甲基-1,3,5-三[β-(二甲基氯硅基)乙基]環三硅氮烷溶液。
(3)
a.將1,1′-烴基硅甲撐-2,2′-雙(二甲基羥基硅基)雙碳硼烷溶解于除水芳烴溶劑中,加入三乙胺,配制成碳硼烷單體溶液,備用。
b.在氮氣保護下,將碳硼烷單體溶液滴加到上述(2)中制備的1,3,5-三甲基-1,3,5-三[β-(二甲基氯硅基)乙基]環三硅氮烷溶液中進行縮聚反應,反應溫度保持在25℃~75℃,反應時間為1~48小時。
c.反應結束后,等待反應液自然冷卻至室溫,過濾除去不溶的三乙胺鹽酸鹽,濾液加入萃取劑萃取,每次加入萃取劑5~30份,共萃取3~5次。
d.合并萃取液,然后用去離子水水洗至中性,加入0.5~20份干燥劑干燥,旋蒸除去萃取劑,得到產物硅基碳硼烷聚合物。
比如上述的硅基碳硼烷聚合物的制備方法,可以如下
(1)
a.按重量計,將0.2~2份氫氧化鈉,20~200份去離子水,混合均勻,再加入0.2~5份一水磷酸二氫鈉,攪拌溶解,制備成緩沖溶液,備用。
b.按重量計,將上述a中配制好的緩沖溶液加入到反應器中,再加入5~50份的1,4-二氧六環以及0.1~2份的貴金屬催化劑,最后加入0.5~5份1,1′-烴基硅甲撐-2,2′-雙(二甲基硅基)雙碳硼烷溶于10~60份芳烴溶劑中所形成的溶液,在溫度為25℃~65℃的條件下,進行羥基化反應,反應時間為1~48小時。
c.反應結束后,過濾除去不溶的貴金屬催化劑,濾液加入萃取劑萃取,每次加入萃取劑5~30份,共萃取3~5次。
d.合并萃取液,然后用去離子水洗至中性,加入1~20份干燥劑干燥30分鐘~8小時,旋蒸除去萃取劑,得到產物,1,1′-烴基硅甲撐-2,2′-雙(二甲基羥基硅基)雙碳硼烷。
(2)
a.按重量計,由0.5~5份二甲基氯硅烷,5~50份除水芳烴溶劑配制的二甲基氯硅烷溶液,備用。
b.按重量計,在反應器中投入0.5~5份三乙烯基三甲基環三硅氮烷和5~50份除水芳烴溶劑,氮氣保護下,加入0.001~0.2份鉑催化劑,再加入上述a中配制好的二甲基氯硅烷溶液。設定反應溫度為25~85℃,在氮氣保護下攪拌反應,反應時間為1~48小時,得到1,3,5-三甲基-1,3,5-三[β-(二甲基氯硅基)乙基]環三硅氮烷。
(3)
a.按重量計,將1~5份1,1′-烴基硅甲撐-2,2′-雙(二甲基羥基硅基)雙碳硼烷溶解于1~50份除水芳烴溶劑中,再加入0.5~10份三乙胺。在氮氣保護下,將該溶液滴加到上述(2)中制備的1,3,5-三甲基-1,3,5-三[β-(二甲基氯硅基)乙基]環三硅氮烷溶液中進行縮聚反應,反應溫度保持在25℃~75℃,反應時間為1~48小時。
b.反應結束后,等待反應液自然冷卻至室溫,過濾除去不溶的三乙胺鹽酸鹽顆粒,濾液加入萃取劑萃取,每次加入萃取劑5~30份,共萃取3~5次。
合并萃取液,然后用去離子水水洗至中性,加入0.5~20份干燥劑干燥,旋蒸除去萃取劑,得到產物硅基碳硼烷聚合物。
本發明中,所述的萃取劑為乙醚,乙酸乙酯,正己烷或其中任意兩者以任意重量比混合的溶劑;所述的干燥劑為無水硫酸鎂,無水硫酸鈉,無水氯化鈣中的任意一種;所述的芳烴溶劑為甲苯,二甲苯或兩者以任意比例混合的混合溶劑;所述的旋蒸除萃取劑的條件為溫度30~60℃,真空度為10~20mmhg。
本發明與現有技術相比的突出優點是:
1.與現有技術中含硅碳硼烷聚合物不同,本發明公開的硅基碳硼烷聚合物分子主鏈上含環狀三硅氮烷單元,該結構單元可賦予預聚物以充分的交聯反應性和交聯程度,有利于后續交聯反應。
2.本發明公開的硅基碳硼烷聚合物由工業化原料環狀三硅氮烷與無毒的碳硼烷交替共聚而成,其中的環狀三硅氮烷具有三個取代反應官能團,由此得到其與二官能度的碳硼烷交替共聚形成交聯網絡結構,該交聯網絡結構非常均勻,這也有利于硅氮元素、硼元素分布更均勻,有利于聚合物作為耐高溫材料的性能發揮。
3.本發明公開的新型硅基碳硼烷聚合物分子結構中具有雙碳硼烷夾心結構,硼元素含量更高,從而擁有更好的耐熱性能和耐熱氧化性能。
4.本發明制備新型硅基碳硼烷聚合物所使用的原材料均為市售原料,無毒安全,來源廣泛,價格便宜。制備反應條件溫和、工藝簡單且產物易于提純,適用工業化生產。
附圖說明
圖1是實施例一制備的1,1′-雙苯基硅甲撐-2,2′-雙(二甲基羥基硅基)雙碳硼烷的紅外吸收曲線圖;
圖2是實施例一制備的1,3,5-三甲基-1,3,5-三[β-(二甲基氯硅基)乙基]環三硅氮烷的紅外吸收曲線圖;
圖3是實施例一制備的新型硅基碳硼烷聚合物的紅外吸收曲線圖;
圖4是實施例一制備的新型硅基碳硼烷聚合物的氫核磁共振光譜圖;
圖5是實施例一、實施例二、實施例三、實施例四中涂層的碳纖維以及未涂層的碳纖維的質量殘留與烘烤時間的關系圖。
具體實施方式
下面結合附圖和實施例對本發明技術方案作進一步的闡述。
實施例一
1.合成1,1′-雙苯基硅甲撐-2,2′-雙(二甲基羥基硅基)雙碳硼烷
在1000ml的三口燒瓶中加入135克除水四氫呋喃和13.2克的二苯基二氯硅烷,搭好實驗裝置后,先通氮氣除去裝置內的空氣,而后通過恒壓滴液漏斗滴加25克的乙炔基溴化鎂格氏試劑,滴加時間為30分鐘,設置反應溫度為40℃,反應時間為4小時,同時配置6.2克癸硼烷,78克乙腈和178克四氫呋喃的混合溶液,反應4小時之后,同樣通過恒壓滴液漏斗滴加,滴加時間為30分鐘。滴加結束之后,調節反應溫度為80℃,反應時間為48小時。
反應結束后,加入配置好的110克乙腈,55克丙酮,36克濃鹽酸和50克去離子水混合溶液進行淬滅,淬滅時間為6小時,直至不再有氣泡產生。淬滅結束后,加入無水乙醚對反應液進行萃取,每次加入200克乙醚,共萃取3次。合并萃取液,分出油層,然后用去離子水洗至中性,加入30克無水硫酸鎂干燥5小時,最后過濾,于45℃、真空度為20mmhg旋蒸至恒重,得到9.8g雙苯基硅甲撐雙碳硼烷。
在100ml三口燒瓶中加入17.82克干燥四氫呋喃和2.36克雙苯基硅甲撐雙碳硼烷,搭好反應裝置后,先通氮氣除去裝置內的空氣,在冰浴的條件下等三口燒瓶中溶液的溫度降至0℃,而后通過恒壓滴液漏斗滴加6.00克1.6m正丁基鋰己烷溶液,滴加時間為15分鐘,反應時間為2小時,保持反應溫度為零下。反應兩個小時后,同樣在冰浴的條件下滴加0.95克二甲基氯硅烷和8.92克干燥四氫呋喃的混合溶液,滴加過程為15分鐘,反應1小時后轉移至室溫的條件下繼續反應,反應時間為24小時。
反應結束后,加入20.58克飽和氯化銨溶液進行淬滅,淬滅時間為12小時。淬滅結束后,用無水乙醚對反應液進行萃取,分液,然后用去離子水洗至中性,4.00克無水硫酸鎂干燥5小時,最后過濾,于35℃、真空度為20mmhg下旋蒸至恒重,得到產物1,1′-雙苯基硅甲撐-2,2′-雙(二甲基硅基)雙碳硼烷。
配緩沖溶液:將0.20克氫氧化鈉加入到50.00g去離子水中配置成0.1mol/l的氫氧化鈉溶液,溶解后加入0.65克的一水磷酸二氫鈉,攪拌溶解。
在250ml的三口燒瓶中先加入緩沖溶液,15.51克的1,4-二氧六環和0.50克的鈀碳催化劑,搭好實驗裝置后,通過恒壓滴液漏斗滴加配置好的3.00克1,1′-雙苯基硅甲撐-2,2′-雙(二甲基硅基)雙碳硼烷和17.50克除水甲苯的混合溶液,滴加時間為5分鐘,設置反應溫度為35℃,反應時間為12小時。
反應結束后,抽濾除去不溶的鈀碳催化劑,濾液用無水乙醚萃取,每次加入20克無水乙醚,共萃取3次。合并萃取液,分出油層,然后用去離子水水洗至中性,加入5克無水硫酸鎂干燥5小時,最后過濾,于55℃,真空度為20mmhg旋蒸至恒重,得到產物1,1′-雙苯基硅甲撐-2,2′-雙(二甲基羥基硅基)雙碳硼烷。
2.合成1,3,5-三甲基-1,3,5-三[β-(二甲基氯硅基)乙基]環三硅氮烷
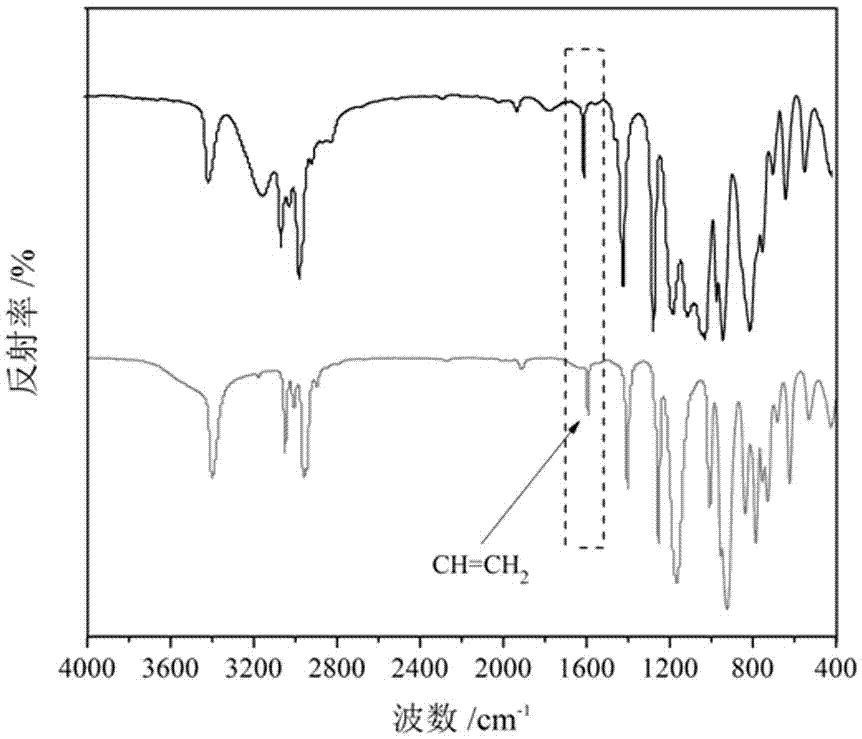
在100ml的三口燒瓶中先加入17.50克除水甲苯溶液,1.27克的三乙烯基三甲基環三硅氮烷,以及0.01克卡斯特催化劑,搭好實驗裝置后,先通氮氣除去裝置內的空氣,而后通過恒壓滴液漏斗滴加配置好的1.42克二甲基氯硅烷和8.75克除水甲苯的混合溶液,滴加時間為5分鐘,在25℃的條件下反應1小時,隨后升溫至75℃反應4小時,反應液變為銀灰色,得到產物1,3,5-三甲基-1,3,5-三[β-(二甲基氯硅基)乙基]環三硅氮烷,直接進入下一步反應。
3.縮聚反應
在100ml的燒杯中加入13.00克的無水甲苯,3.07克1,1′-雙苯基硅甲撐-2,2′-雙(二甲基羥基硅基)雙碳硼烷,以及1.02克的三乙胺,混合均勻,備用。
在上述步驟2得到的1,3,5-三甲基-1,3,5-三[β-(二甲基氯硅基)乙基]環三硅氮烷中,通過恒壓滴液漏斗滴加配制好的碳硼烷單體溶液,滴加時間為15分鐘,在室溫下反應30分鐘后,升溫至50℃,反應時間為18小時。
反應結束后,自然冷卻至室溫,抽濾除去不溶的三乙胺鹽酸鹽,濾液用25ml無水乙醚萃取3次,分液,然后用去離子水水洗至中性,5克無水硫酸鎂干燥5小時,最后過濾,在55℃、真空度為20mmhg下旋蒸至恒重,得到產物,一種新型的硅基碳硼烷聚合物。
參見附圖1,是按本實施例技術方案制備的1,1′-雙苯基硅甲撐-2,2′-雙(二甲基羥基硅基)雙碳硼烷的紅外吸收曲線圖譜。從圖中可以看到,2129.13cm-1處為si-h的伸縮振動峰消失以及843.15cm-1處為si-oh的伸縮振動峰的出現,可以有效的證明了硅羥基化反應的進行。
參見附圖2,是按本實施例技術方案制備的1,3,5-三甲基-1,3,5-三[β-(二甲基氯硅基)乙基]環三硅氮烷的紅外吸收曲線圖譜。從圖中可以看到,1594.83cm-1處為c=ch2的伸縮振動峰減弱,還留有部分的c=ch2峰是由于反應不完全,但也足以證明了硅氫加成反應的發生。
參見附圖3,是按本實施例技術方案制備的新型硅基碳硼烷聚合物的紅外吸收曲線圖譜。從圖中可以看到,3379.83cm-1處的吸收峰是n-h的伸縮振動峰;碳硼烷的b-h峰在2563.46cm-1處;而843.15cm-1處為si-oh的伸縮振動峰的消失以及1031.58cm-1處si-o-sicm-1伸縮振動峰的出現則有力的證實了縮聚反應的發生。
參見附圖4,是按本實施例技術方案制備的新型硅基碳硼烷聚合物氫核磁共振光譜。從圖中可以看到,0.05-0.26(m,si-ch3);0.4-2.0(br,b-h);7.2-7.7(d,ph-h);2.27(d,n-h);5.86-6.08(m,ch=ch2);結合紅外吸收圖譜,證實了聚合物的合成。
產物分子結構式如下:
式中,m和n分別4~8的整數
4.涂層處理
將硅基碳硼烷聚合物溶解在二氯甲烷溶液中,配制成質量濃度為0.12g/毫升的溶液,備用。將t-300碳纖維在氮氣保護和400℃的條件下烘烤2小時,再在質量濃度為5%的硝酸溶液中浸泡1小時,然后去離子水淋洗至中性。再將處理好的碳纖維浸泡在硅基碳硼烷聚合物的二氯甲烷溶液中,室溫自然風干。
重復處理3次,得到表面有3層sibcn陶瓷涂層的碳纖維,碳纖維的拉伸強度為3982mpa。
測試了處理前后碳纖維的耐熱穩定性能,結果見附圖5。從圖中可以看出,未經處理的碳纖維在500℃的條件下,80分鐘便已經完全降解,而經過3次涂層處理的碳纖維,即使加熱至900℃的條件下烘烤2小時后,質量殘留高達99%。
實施例二
1.合成1,1′-雙苯基硅甲撐-2,2′-雙(二甲基羥基硅基)雙碳硼烷
在1000ml的三口燒瓶中加入135克除水四氫呋喃和13.2克的二苯基二氯硅烷,搭好實驗裝置后,先通氮氣除去裝置內的空氣,而后通過恒壓滴液漏斗滴加25克的乙炔基溴化鎂格氏試劑,滴加時間為30分鐘,設置反應溫度為40℃,反應時間為4小時,同時配置6.2克癸硼烷,78克乙腈和178克四氫呋喃的混合溶液,反應4小時之后,同樣通過恒壓滴液漏斗滴加,滴加時間為30分鐘。滴加結束之后,調節反應溫度為86℃,反應時間為48小時。
反應結束后,加入配置好的110克乙腈,55克丙酮,36克濃鹽酸和50克去離子水混合溶液進行淬滅,淬滅時間為6小時,直至不再有氣泡產生。淬滅結束后,加入無水乙醚對反應液進行萃取,每次加入200克乙醚,共萃取3次。合并萃取液,分出油層,然后用去離子水洗至中性,加入30克無水硫酸鎂干燥5小時,最后過濾,于45℃、真空度為20mmhg旋蒸至恒重,得到9.8g雙苯基硅甲撐雙碳硼烷。
在250ml三口燒瓶中加入35.64克干燥四氫呋喃和4.75克雙苯基硅甲撐雙碳硼烷,搭好實驗裝置后,先通氮氣除去裝置內的空氣,在冰浴的條件下等三口燒瓶中溶液的溫度降至0℃而后通過恒壓滴液漏斗滴加12.50克1.6m正丁基鋰己烷溶液,滴加時間為20分鐘,反應時間為4小時,保持反應溫度為零下。反應4個小時后,同樣在冰浴的條件下滴加1.92克二甲基氯硅烷和17.85克干燥四氫呋喃的混合溶液,滴加過程為20分鐘,反應1小時后轉移至室溫的條件下繼續反應,反應時間為24小時。
反應結束后,加入41.16克飽和氯化銨溶液進行淬滅,淬滅時間為20小時。淬滅結束后,用無水乙醚對反應液進行萃取,分液,然后用去離子水洗至中性,10克無水硫酸鎂干燥5小時,最后過濾,于35℃、真空度為20mmhg下旋蒸至恒重,得到產物1,1′-雙苯基硅甲撐-2,2′-雙(二甲基硅基)雙碳硼烷。
配置緩沖溶液:將0.40克氫氧化鈉加入到100.00g去離子水中配置成0.10mol/l的氫氧化鈉溶液,溶解后加入1.30克的一水磷酸二氫鈉,攪拌溶解。
在250ml的三口燒瓶中先加入緩沖溶液,32.00克1,4-二氧六環和1.50克的鈀碳催化劑,搭好實驗裝置后,通過恒壓滴液漏斗滴加配置好的6.00克1,1′-雙苯基硅甲撐-2,2′-雙(二甲基硅基)雙碳硼烷和35.50克除水甲苯的混合溶液,滴加時間為5分鐘,設置反應溫度為35℃,反應時間為12小時。
反應結束后,抽濾除去不溶的鈀碳催化劑,濾液用無水乙醚萃取,每次加入40克無水乙醚,共萃取5次。合并萃取液,分出油層,然后用去離子水水洗至中性,加入15克無水硫酸鎂干燥5小時,最后過濾,于60℃,真空度為20mmhg旋蒸至恒重,得到產物1,1′-雙苯基硅甲撐-2,2′-雙(二甲基羥基硅基)雙碳硼烷。
2.合成1,3,5-三甲基-1,3,5-三[β-(二甲基氯硅基)乙基]環三硅氮烷
在250ml的三口燒瓶中先加入35.00克除水甲苯溶液,2.54克的三乙烯基三甲基環三硅氮烷,以及0.02克卡斯特催化劑,搭好實驗裝置后,先通氮氣除去裝置內的空氣,而后通過恒壓滴液漏斗滴加配置好的2.84克二甲基氯硅烷和17.75克除水甲苯的混合溶液,滴加時間為10分鐘,在35℃的條件下反應1小時,隨后升溫至75℃反應4小時,反應液變為銀灰色,得到產物1,3,5-三甲基-1,3,5-三[β-(二甲基氯硅基)乙基]環三硅氮烷,直接進入下一步反應。
3.縮聚反應
在250ml的燒杯中加入26.00克的無水甲苯,6.54克1,1′-雙苯基硅甲撐-2,2′-雙(二甲基羥基硅基)雙碳硼烷,以及2.04克的三乙胺,混合均勻,備用。
在上述步驟2得到的1,3,5-三甲基-1,3,5-三[β-(二甲基氯硅基)乙基]環三硅氮烷中,通過恒壓滴液漏斗滴加配制好的碳硼烷單體溶液,滴加時間為30分鐘,在室溫下反應1小時后,升溫至55℃,反應時間為24小時。
反應結束后,自然冷卻至室溫,抽濾除去不溶的三乙胺鹽酸鹽,濾液用無水乙醚萃取,分液,然后用去離子水水洗至中性,10克無水硫酸鎂干燥5小時,最后過濾,在55℃、真空度為15mmhg下旋蒸至恒重,得到產物硅基碳硼烷聚合物。
m和n分別為5~10的整數
4.涂層處理
將硅基碳硼烷聚合物溶解在二氯甲烷溶液中,配制成質量濃度為0.36g/毫升的溶液,備用。
將t-300碳纖維在氮氣保護和400℃的條件下烘烤2小時,再在質量濃度為5%的硝酸溶液中浸泡1小時,然后去離子水淋洗至中性。再將碳纖維浸泡在硅基碳硼烷聚合物的二氯甲烷溶液中,室溫自然風干。重復處理3次得到表面有3層涂層的碳纖維,測試了處理前后碳纖維的耐熱穩定性能,結果見附圖5。加熱至700℃的條件下烘烤2小時,測得處理后碳纖維質量殘余70.39%。
實施例三
1.合成1,1′-雙苯基硅甲撐-2,2′-雙(二甲基羥基硅基)雙碳硼烷
合成雙苯基硅甲撐雙碳硼烷同實施例一。
在1000ml三口燒瓶中加入71.28克干燥四氫呋喃和9.55克雙苯基硅甲撐雙碳硼烷,搭好實驗裝置后,先通氮氣除去裝置內的空氣,在冰浴的條件下等三口燒瓶中溶液的溫度降至0℃而后通過恒壓滴液漏斗滴加25.50克1.6m正丁基鋰己烷溶液,滴加時間為60分鐘,反應時間為8小時,保持反應溫度為零下。反應8個小時后,同樣在冰浴的條件下滴加3.95克二甲基氯硅烷和35.75克干燥四氫呋喃的混合溶液,滴加過程為30分鐘,反應2小時后轉移至室溫的條件下繼續反應,反應時間為24小時。
反應結束后,加入82.32克飽和氯化銨溶液進行淬滅,淬滅時間為20小時。淬滅結束后,用無水乙醚對反應液進行萃取,分液,然后用去離子水洗至中性,20克無水硫酸鎂干燥5小時,最后過濾,于40℃、真空度為20mmhg下旋蒸至恒重,得到產物1,1′-雙苯基硅甲撐-2,2′-雙(二甲基硅基)雙碳硼烷。
配制緩沖溶液:將0.80克氫氧化鈉加入到200.00g去離子水中配置成0.10mol/l的氫氧化鈉溶液,溶解后加入2.60克的一水磷酸二氫鈉,攪拌溶解。
在250ml的三口燒瓶中先加入緩沖溶液,64.00克1,4-二氧六環和3.50克的鈀碳催化劑,搭好實驗裝置后,通過恒壓滴液漏斗滴加配置好的12.50克1,1′-雙苯基硅甲撐-2,2′-雙(二甲基硅基)雙碳硼烷和71.50克除水甲苯的混合溶液,滴加時間為30分鐘,設置反應溫度為55℃,反應時間為12小時。
反應結束后,抽濾除去不溶的鈀碳催化劑,濾液用無水乙醚萃取,每次加入100克無水乙醚,共萃取3次。合并萃取液,分出油層,然后用去離子水水洗至中性,加入50克無水硫酸鎂干燥5小時,最后過濾,于55℃,真空度為20mmhg旋蒸至恒重,得到產物1,1′-雙苯基硅甲撐-2,2′-雙(二甲基羥基硅基)雙碳硼烷。
2.合成1,3,5-三甲基-1,3,5-三[β-(二甲基氯硅基)乙基]環三硅氮烷
在1000ml的三口燒瓶中先加入75.50克除水甲苯溶液,5.25克的三乙烯基三甲基環三硅氮烷,以及0.05克卡斯特催化劑,搭好實驗裝置后,先通氮氣除去裝置內的空氣,而后通過恒壓滴液漏斗滴加配置好的5.75克二甲基氯硅烷和35.50克除水甲苯的混合溶液,滴加時間為30分鐘,在45℃的條件下反應1小時,隨后升溫至75℃反應5小時,反應液變為銀灰色,得到產物1,3,5-三甲基-1,3,5-三[β-(二甲基氯硅基)乙基]環三硅氮烷,直接進入下一步反應。
3.縮聚反應
在1000ml的燒杯中加入55.00克的無水甲苯,6.50克1,1′-雙苯基硅甲撐-2,2′-雙(二甲基羥基硅基)雙碳硼烷,以及2.50克的三乙胺,混合均勻,得到碳硼烷單體溶液,備用。
在上述步驟2得到的1,3,5-三甲基-1,3,5-三[β-(二甲基氯硅基)乙基]環三硅氮烷中,通過恒壓滴液漏斗滴加配制好的碳硼烷單體溶液,滴加時間為45分鐘,在室溫下反應120分鐘后,升溫至55℃,反應時間為36小時。
反應結束后,自然冷卻至室溫,抽濾除去不溶的三乙胺鹽酸鹽,濾液用無水乙醚萃取,分液,然后用去離子水水洗至中性,50克無水硫酸鎂干燥12小時,最后過濾,在55℃、真空度為20mmhg下旋蒸至恒重,得到產物,一種新型的硅基碳硼烷聚合物。
產物分子結構式如下:
式中,m和n分別4~6的整數
4.涂層處理
將硅基碳硼烷聚合物溶解在二氯甲烷溶液中,配制成質量濃度為0.12g/毫升的溶液,備用。將t-300碳纖維在氮氣保護和400℃的條件下烘烤2小時,再在質量濃度為5%的硝酸溶液中浸泡1小時,然后去離子水淋洗至中性。再將處理好的碳纖維浸泡在碳硼烷陶瓷前驅體的二氯甲烷溶液中,室溫自然風干。重復處理3次得到表面有3層涂層的碳纖維t-300。
測試了處理前后碳纖維的耐熱穩定性能,結果見附圖5。將處理后碳纖維加熱至800℃烘烤2小時,發現處理后碳纖維在800℃下氧化60分鐘以后質量保持恒定,且碳纖維質量殘余76.65%。
實施例四
1.合成1,1′-雙苯基硅甲撐-2,2′-雙(二甲基羥基硅基)雙碳硼烷
在2000ml三口燒瓶中加入145.50克干燥四氫呋喃和19.32克雙苯基硅甲撐雙碳硼烷(實施例二),搭好實驗裝置后,先通氮氣除去裝置內的空氣,在冰浴的條件下等三口燒瓶中溶液的溫度降至0℃而后通過恒壓滴液漏斗滴加55.25克1.6m正丁基鋰己烷溶液,滴加時間為120分鐘,反應時間為8小時,保持反應溫度為零下。反應8個小時后,同樣在冰浴的條件下滴加7.68克二甲基氯硅烷和71.45克干燥四氫呋喃的混合溶液,滴加過程為60分鐘,反應2小時后轉移至室溫的條件下繼續反應,反應時間為36小時。
反應結束后,加入164.64克飽和氯化銨溶液進行淬滅,淬滅時間為20小時。淬滅結束后,用無水乙酸乙酯對反應液進行萃取,分液,然后用去離子水洗至中性,40克無水硫酸鎂干燥12小時,最后過濾,于35℃、真空度為10mmhg下旋蒸至恒重,得到產物1,1′-雙苯基硅甲撐-2,2′-雙(二甲基硅基)雙碳硼烷。
配制緩沖溶液:將1.60克氫氧化鈉加入到400.00g去離子水中配置成0.10mol/l的氫氧化鈉溶液,溶解后,加入5.20克的一水磷酸二氫鈉,攪拌溶解。
在2000ml的三口燒瓶中先加入緩沖溶液,135.50克的1,4-二氧六環和6.50克的鈀碳催化劑,搭好實驗裝置后,通過恒壓滴液漏斗滴加配置好的24.30克1,1′-雙苯基硅甲撐-2,2′-雙(二甲基硅基)雙碳硼烷和142.50克除水甲苯的混合溶液,滴加時間為5分鐘,設置反應溫度為35℃,反應時間為12小時。
反應結束后,抽濾除去不溶的鈀碳催化劑,濾液用無水正己烷萃取,每次加入40克無水正己烷,共萃取5次。合并萃取液,分出油層,然后用去離子水水洗至中性,加入100克無水硫酸鎂干燥12小時,最后過濾,于55℃,真空度為15mmhg旋蒸至恒重,得到產物1,1′-雙苯基硅甲撐-2,2′-雙(二甲基羥基硅基)雙碳硼烷。
2.合成1,3,5-三甲基-1,3,5-三[β-(二甲基氯硅基)乙基]環三硅氮烷
在2000ml的三口燒瓶中先加入145.00克除水甲苯溶液,10.50克的三乙烯基三甲基環三硅氮烷,以及0.10克卡斯特催化劑,搭好實驗裝置后,先通氮氣除去裝置內的空氣,而后通過恒壓滴液漏斗滴加配置好的11.52克二甲基氯硅烷和71.50克除水甲苯的混合溶液,滴加時間為120分鐘,在35℃的條件下反應2小時,隨后升溫至75℃反應10小時,反應液變為銀灰色,得到產物1,3,5-三甲基-1,3,5-三[β-(二甲基氯硅基)乙基]環三硅氮烷,直接進入下一步反應。
3.縮聚反應
在2000ml的燒杯中加入105.50克的無水甲苯,27.80克1,1′-雙苯基硅甲撐-2,2′-雙(二甲基羥基硅基)雙碳硼烷,以及8.53克的三乙胺,混合均勻,備用。
在上述步驟2得到的1,3,5-三甲基-1,3,5-三[β-(二甲基氯硅基)乙基]環三硅氮烷中,通過恒壓滴液漏斗滴加配制好的碳硼烷單體溶液,滴加時間為120分鐘,在室溫下反應4小時后,升溫至55℃,反應時間為48小時。
反應結束后,自然冷卻至室溫,抽濾除去不溶的三乙胺鹽酸鹽,濾液用無水正己烷萃取,分液,然后用去離子水水洗至中性,100克無水硫酸鈉干燥5小時,最后過濾,在55℃、真空度為15mmhg下旋蒸至恒重,得到產物,一種新型的硅基碳硼烷聚合物。
產物分子結構式如下:
m和n分別為6~12的整數
4.涂層處理
將碳硼烷陶瓷前驅體溶解在二氯甲烷溶液中,配制成質量濃度為0.12g/毫升的溶液,備用。將碳纖維在氮氣保護和400℃的條件下烘烤2小時,再在質量濃度為5%的硝酸溶液中浸泡1小時,然后去離子水淋洗至中性。再將處理好的碳纖維浸泡在碳硼烷陶瓷前驅體的二氯甲烷溶液中,室溫自然風干。重復處理3次得到有3層涂層的碳纖維t-300。碳纖維加熱至600℃烘烤2小時,質量殘留高于80%,熱穩定性優于未處理碳纖維(見附圖5)。
實施例五
1.合成1,1′-甲基苯基硅甲撐-2,2′-雙(二甲基羥基硅基)雙碳硼烷
在1000ml的三口燒瓶中加入135克除水四氫呋喃和9.6克的甲基苯基二氯硅烷,搭好實驗裝置后,先通氮氣除去裝置內的空氣,而后通過恒壓滴液漏斗滴加22克的乙炔基溴化鎂格氏試劑,滴加時間為35分鐘,設置反應溫度為40℃,反應時間為4小時,同時配置6.2克癸硼烷,78克乙腈和178克四氫呋喃的混合溶液,反應4小時之后,同樣通過恒壓滴液漏斗滴加,滴加時間為35分鐘。滴加結束之后,調節反應溫度為80℃,反應時間為48小時。
反應結束后,加入配置好的110克乙腈,55克丙酮,36克濃鹽酸和50克去離子水混合溶液進行淬滅,淬滅時間為6小時,直至不再有氣泡產生,淬滅結束后,用無水乙醚對反應液進行萃取,分液,然后用去離子水洗至中性,無水硫酸鎂干燥5小時,最后過濾,于30℃、真空度為15mmhg旋蒸至恒重,得到產物甲基苯基硅甲撐雙碳硼烷8.8g。
在100ml三口燒瓶中加入17.82克干燥四氫呋喃和2.36克甲基苯基硅甲撐雙碳硼烷,搭好實驗裝置后,先通氮氣除去裝置內的空氣,在冰浴的條件下等三口燒瓶中溶液的溫度降至0℃,而后通過恒壓滴液漏斗滴加6.00克1.6m正丁基鋰己烷溶液,滴加時間為15分鐘,反應時間為2小時,保持反應溫度為零下。反應兩個小時后,同樣在冰浴的條件下滴加0.95克二甲基氯硅烷和8.92克干燥四氫呋喃的混合溶液,滴加過程為15分鐘,反應1小時后轉移至室溫的條件下繼續反應,反應時間為24小時。
反應結束后,加入20.58克飽和氯化銨溶液進行淬滅,淬滅時間為12小時。淬滅結束后,用無水乙醚對反應液進行萃取,分液,然后用去離子水洗至中性,4.00克無水硫酸鎂干燥5小時,最后過濾,于35℃、真空度為20mmhg下旋蒸至恒重,得到產物1,1′-甲基苯基硅甲撐-2,2′-雙(二甲基硅基)雙碳硼烷。
配制緩沖溶液:將0.20克氫氧化鈉加入到50.00g去離子水中配置成0.1mol/l的氫氧化鈉溶液,溶解后加入0.65克的一水磷酸二氫鈉,攪拌溶解。
在250ml的三口燒瓶中先加入緩沖溶液,15.51克的1,4-二氧六環和0.50
克的鈀碳催化劑,搭好實驗裝置后,通過恒壓滴液漏斗滴加配置好的3.00克1,1′-甲基苯基硅甲撐-2,2′-雙(二甲基硅基)雙碳硼烷和17.50克除水甲苯的混合溶液,滴加時間為5分鐘,設置反應溫度為35℃,反應時間為12小時。
反應結束后,抽濾除去不溶的鈀碳催化劑,濾液用無水乙酸乙酯萃取,每次加入20克無水乙酸乙酯,共萃取3次。合并萃取液,分出油層,然后用去離子水水洗至中性,加入5克無水氯化鈣干燥5小時,最后過濾,于55℃,真空度為20mmhg旋蒸至恒重,得到產物1,1′-甲基苯基硅甲撐-2,2′-雙(二甲基羥基硅基)雙碳硼烷。
2.合成1,3,5-三甲基-1,3,5-三[β-(二甲基氯硅基)乙基]環三硅氮烷
在100ml的三口燒瓶中先加入17.50克除水甲苯溶液,1.27克的三乙烯基三甲基環三硅氮烷,以及0.01克卡斯特催化劑,搭好實驗裝置后,先通氮氣除去裝置內的空氣,而后通過恒壓滴液漏斗滴加配置好的1.42克二甲基氯硅烷和8.75克除水甲苯的混合溶液,滴加時間為5分鐘,在25℃的條件下反應1小時,隨后升溫至75℃反應4小時,反應液變為銀灰色,得到產物1,3,5-三甲基-1,3,5-三[β-(二甲基氯硅基)乙基]環三硅氮烷,直接進入下一步反應。
3.縮聚反應
在100ml的燒杯中加入13.00克的無水甲苯,3.67克1,1′-甲基苯基硅甲撐-2,2′-雙(二甲基羥基硅基)雙碳硼烷,以及1.02克的三乙胺,混合均勻,備用。
在上述步驟2得到的1,3,5-三甲基-1,3,5-三[β-(二甲基氯硅基)乙基]環三硅氮烷中,通過恒壓滴液漏斗滴加配制好的碳硼烷單體溶液,滴加時間為15分鐘,在室溫下反應30分鐘后,升溫至50℃,反應時間為18小時。
反應結束后,自然冷卻至室溫,抽濾除去不溶的三乙胺鹽酸鹽,濾液用無水乙醚萃取,分液,然后用去離子水水洗至中性,5克無水硫酸鎂干燥5小時,最后過濾,在55℃、真空度為20mmhg下旋蒸至恒重,得到產物,一種新型的硅基碳硼烷聚合物。
產物分子結構式如下:
m和n分別為5~12的整數
4.涂層處理
將碳硼烷陶瓷前驅體溶解在三氯甲烷溶液中,配制成質量濃度為0.12g/毫升的溶液,備用。將t-700碳纖維在氮氣保護和400℃的條件下烘烤2小時,再在質量濃度為5%的硝酸溶液中浸泡1小時,然后去離子水淋洗至中性。再將處理好的碳纖維浸泡在碳硼烷陶瓷前驅體的二氯甲烷溶液中,室溫自然風干。重復處理2次得到有2層涂層的碳纖維材料。碳纖維在空氣的氛圍中加熱至1000℃質量保留率達76.24%。
實施例六
1.合成1,1′-甲基苯基硅甲撐-2,2′-雙(二甲基羥基硅基)雙碳硼烷
甲基苯基硅甲撐雙碳硼烷同實施例五。
在250ml三口燒瓶中加入35.64克干燥四氫呋喃和4.72克甲基苯基硅甲撐雙碳硼烷,搭好實驗裝置后,先通氮氣除去裝置內的空氣,在冰浴的條件下等三口燒瓶中溶液的溫度降至0℃,而后通過恒壓滴液漏斗滴加12.50克1.6m正丁基鋰己烷溶液,滴加時間為30分鐘,反應時間為3小時,保持反應溫度為零下。反應3個小時后,同樣在冰浴的條件下滴加1.95克二甲基氯硅烷和17.85克干燥四氫呋喃的混合溶液,滴加過程為30分鐘,反應2小時后轉移至室溫的條件下繼續反應,反應時間為18小時。
反應結束后,加入41.16克飽和氯化銨溶液進行淬滅,淬滅時間為12小時。淬滅結束后,用無水乙酸乙酯對反應液進行萃取,分液,然后用去離子水洗至中性,10.00克無水硫酸鎂干燥5小時,最后過濾,于40℃、真空度為20mmhg下旋蒸至恒重,得到產物1,1′-甲基苯基硅甲撐-2,2′-雙(二甲基硅基)雙碳硼烷。
配置緩沖溶液:將0.40克氫氧化鈉加入到100.00g去離子水中配置成0.1mol/l的氫氧化鈉溶液,溶解后加入1.30克的一水磷酸二氫鈉,攪拌溶解。
在250ml的三口燒瓶中先加入緩沖溶液,32.50克的1,4-二氧六環和1.50
克的鈀碳催化劑,搭好實驗裝置后,通過恒壓滴液漏斗滴加配置好的6.50克1,1′-甲基苯基硅甲撐-2,2′-雙(二甲基硅基)雙碳硼烷和35.50克除水甲苯的混合溶液,滴加時間為15分鐘,設置反應溫度為35℃,反應時間為12小時。
反應結束后,抽濾除去不溶的鈀碳催化劑,濾液用無水乙酸乙酯萃取,每次
加入20克無水乙酸乙酯,共萃取5次。合并萃取液,分出油層,然后用去離子水水洗至中性,加入10克無水氯化鈣干燥5小時,最后過濾,于55℃,真空度為15mmhg旋蒸至恒重,得到產物1,1′-甲基苯基硅甲撐-2,2′-雙(二甲基羥基硅基)雙碳硼烷。
2.合成1,3,5-三甲基-1,3,5-三[β-(二甲基氯硅基)乙基]環三硅氮烷
在250ml的三口燒瓶中先加入35.50克除水甲苯溶液,2.52克的三乙烯基三甲基環三硅氮烷,以及0.04克卡斯特催化劑,搭好實驗裝置后,先通氮氣除去裝置內的空氣,而后通過恒壓滴液漏斗滴加配置好的2.85克二甲基氯硅烷和16.50克除水甲苯的混合溶液,滴加時間為15分鐘,在35℃的條件下反應1小時,隨后升溫至65℃反應5小時,反應液變為銀灰色,得到產物1,3,5-三甲基-1,3,5-三[β-(二甲基氯硅基)乙基]環三硅氮烷,直接進入下一步反應。
3.縮聚反應
在250ml的燒杯中加入26.50克的無水甲苯,6.81克1,1′-甲基苯基硅甲撐-2,2′-雙(二甲基羥基硅基)雙碳硼烷,以及2.42克的三乙胺,混合均勻,備用。
在上述步驟2得到的1,3,5-三甲基-1,3,5-三[β-(二甲基氯硅基)乙基]環三硅氮烷中,通過恒壓滴液漏斗滴加配制好的碳硼烷單體溶液,滴加時間為20分鐘,在室溫下反應50分鐘后,升溫至50℃,反應時間為24小時。
反應結束后,自然冷卻至室溫,抽濾除去不溶的三乙胺鹽酸鹽,濾液用無水正己烷萃取,分液,然后用去離子水水洗至中性,10克無水硫酸鈉干燥5小時,最后過濾,在55℃、真空度為20mmhg下旋蒸至恒重,得到產物,一種新型的硅基碳硼烷聚合物。產物分子結構式如下:
m和n分別為6~12的整數
4.涂層處理
將碳硼烷陶瓷前驅體溶解在二氯乙烷溶液中,配制成質量濃度為0.12g/毫升的溶液,備用。將t900碳纖維在氮氣保護和400℃的條件下烘烤2小時,再在質量濃度為5%的硝酸溶液中浸泡1小時,然后去離子水淋洗至中性。再將處理好的碳纖維浸泡在碳硼烷陶瓷前驅體的二氯甲烷溶液中,室溫自然風干。重復處理3次得到有3層涂層的碳纖維材料。處理后碳纖維在空氣的氛圍中加熱至1000℃質量保留率為75.57%。
實施例七
1.合成1,1′-雙苯基硅甲撐-2,2′-雙(二甲基羥基硅基)雙碳硼烷
在2000ml三口燒瓶中加入132.68克干燥四氫呋喃和19.30克雙苯基硅甲撐雙碳硼烷(實施例二),搭好實驗裝置后,先通氮氣除去裝置內的空氣,在冰浴的條件下等三口燒瓶中溶液的溫度降至0℃而后通過恒壓滴液漏斗滴加55.29克1.6m正丁基鋰己烷溶液,滴加時間為120分鐘,反應時間為8小時,保持反應溫度為零下。反應8個小時后,同樣在冰浴的條件下滴加7.67克二甲基氯硅烷和71.40克干燥四氫呋喃的混合溶液,滴加過程為60分鐘,反應2小時后轉移至室溫的條件下繼續反應,反應時間為36小時。
反應結束后,加入164.23克飽和氯化銨溶液進行淬滅,淬滅時間為20小時。淬滅結束后,用無水乙酸乙酯對反應液進行萃取,分液,然后用去離子水洗至中性,40克無水硫酸鎂干燥12小時,最后過濾,于35℃、真空度為10mmhg下旋蒸至恒重,得到產物1,1′-雙苯基硅甲撐-2,2′-雙(二甲基硅基)雙碳硼烷。
配緩沖溶液:將1.60克氫氧化鈉加入到400.00g去離子水中配置成0.10mol/l的氫氧化鈉溶液,溶解后加入5.20克的一水磷酸二氫鈉,攪拌溶解。
在2000ml的三口燒瓶中先加入緩沖溶液,135.12克的1,4-二氧六環和6.35克的鈀碳催化劑,搭好實驗裝置后,通過恒壓滴液漏斗滴加配置好的24.12克1,1′-雙苯基硅甲撐-2,2′-雙(二甲基硅基)雙碳硼烷和142.50克除水甲苯的混合溶液,滴加時間為5分鐘,設置反應溫度為35℃,反應時間為12小時。
反應結束后,抽濾除去不溶的鈀碳催化劑,濾液用無水正己烷萃取,每次加入40克無水正己烷,共萃取5次。合并萃取液,分出油層,然后用去離子水水洗至中性,加入100克無水硫酸鎂干燥12小時,最后過濾,于55℃,真空度為15mmhg旋蒸至恒重,得到產物1,1′-雙苯基硅甲撐-2,2′-雙(二甲基羥基硅基)雙碳硼烷。
2.合成1,3,5-三甲基-1,3,5-三[β-(二甲基氯硅基)乙基]環三硅氮烷
在2000ml的三口燒瓶中先加入145克除水甲苯溶液,10.34克的三乙烯基三甲基環三硅氮烷,以及0.10克卡斯特催化劑,搭好實驗裝置后,先通氮氣除去裝置內的空氣,而后通過恒壓滴液漏斗滴加配置好的11.42克二甲基氯硅烷和71.50克除水甲苯的混合溶液,滴加時間為120分鐘,在35℃的條件下反應2小時,隨后升溫至75℃反應10小時,反應液變為銀灰色,得到產物1,3,5-三甲基-1,3,5-三[β-(二甲基氯硅基)乙基]環三硅氮烷,直接進入下一步反應。
3.縮聚反應
在2000ml的燒杯中加入105.50克的無水甲苯,24.56克1,1′-雙苯基硅甲撐-2,2′-雙(二甲基羥基硅基)雙碳硼烷,以及8.53克的三乙胺,混合均勻,備用。
在上述步驟2得到的1,3,5-三甲基-1,3,5-三[β-(二甲基氯硅基)乙基]環三硅氮烷中,通過恒壓滴液漏斗滴加配制好的碳硼烷單體溶液,滴加時間為120分鐘,在室溫下反應4小時后,升溫至55℃,反應時間為48小時。
反應結束后,自然冷卻至室溫,抽濾除去不溶的三乙胺鹽酸鹽,濾液用無水正己烷萃取,分液,然后用去離子水水洗至中性,100克無水硫酸鈉干燥5小時,最后過濾,在55℃、真空度為15mmhg下旋蒸至恒重,得到產物為一種新型的碳硼烷陶瓷前驅體。
碳硼烷陶瓷前驅體分子結構式如下:
m和n分別為5~10的整數
4.涂層處理
將碳硼烷陶瓷前驅體溶解在二氯乙烷溶液中,配制成質量濃度為0.12g/毫升的溶液,備用。將碳纖維在氮氣保護和400℃的條件下烘烤2小時,再在質量濃度為5%的硝酸溶液中浸泡1小時,然后去離子水淋洗至中性。再將處理好的碳纖維浸泡在碳硼烷陶瓷前驅體的二氯乙烷溶液中,室溫自然風干,得到單層涂層的碳纖維t-300。碳纖維加熱至600℃烘烤2小時,質量保留率為34%,熱穩定性優于未處理碳纖維。涂層處理后,碳纖維的拉伸強度為4728mpa,處理后拉伸強度僅損失3.50%。
實施例八
1.合成1,1′-甲基苯基硅甲撐-2,2′-雙(二甲基羥基硅基)雙碳硼烷
在1000ml的三口燒瓶中加入150克除水四氫呋喃和9.6克的甲基苯基二氯硅烷,搭好實驗裝置后,先通氮氣除去裝置內的空氣,而后通過恒壓滴液漏斗滴加22克的乙炔基溴化鎂格氏試劑,滴加時間為35分鐘,設置反應溫度為40°c,反應時間為4小時,同時配置6.2克癸硼烷,78克乙腈和178克四氫呋喃的混合溶液,反應4小時之后,同樣通過恒壓滴液漏斗滴加,滴加時間為35分鐘。滴加結束之后,調節反應溫度為86℃,反應時間為48小時。
反應結束后,加入配置好的110克乙腈,55克丙酮,36克濃鹽酸和50克去離子水混合溶液進行淬滅,淬滅時間為6小時,直至不再有氣泡產生,淬滅結束后,用無水乙醚對反應液進行萃取,分液,然后用去離子水洗至中性,無水硫酸鎂干燥5小時,最后過濾,于30℃、真空度為15mmhg旋蒸至恒重,得到產物甲基苯基硅甲撐雙碳硼烷8.8g。
在100ml三口燒瓶中加入17.82克干燥四氫呋喃和2.36克甲基苯基硅甲撐雙碳硼烷,搭好實驗裝置后,先通氮氣除去裝置內的空氣,在冰浴的條件下等三口燒瓶中溶液的溫度降至0℃,而后通過恒壓滴液漏斗滴加6.00克1.6m正丁基鋰己烷溶液,滴加時間為15分鐘,反應時間為2小時,保持反應溫度為零下。反應兩個小時后,同樣在冰浴的條件下滴加0.95克二甲基氯硅烷和8.92克干燥四氫呋喃的混合溶液,滴加過程為15分鐘,反應1小時后轉移至室溫的條件下繼續反應,反應時間為24小時。
反應結束后,加入20.58克飽和氯化銨溶液進行淬滅,淬滅時間為12小時。淬滅結束后,用無水乙醚對反應液進行萃取,分液,然后用去離子水洗至中性,4.00克無水硫酸鎂干燥5小時,最后過濾,于35℃、真空度為20mmhg下旋蒸至恒重,得到產物1,1′-甲基苯基硅甲撐-2,2′-雙(二甲基硅基)雙碳硼烷。
配制緩沖溶液:將0.20克氫氧化鈉加入到50.00g去離子水中配置成0.1mol/l的氫氧化鈉溶液,溶解后加入0.65克的一水磷酸二氫鈉,攪拌溶解。
在250ml的三口燒瓶中先加入緩沖溶液,15.51克的1,4-二氧六環和0.50克的鈀碳催化劑,搭好實驗裝置后,通過恒壓滴液漏斗滴加配置好的3.00克1,1′-甲基苯基硅甲撐-2,2′-雙(二甲基硅基)雙碳硼烷和17.50克除水甲苯的混合溶液,滴加時間為5分鐘,設置反應溫度為35℃,反應時間為12小時。
反應結束后,抽濾除去不溶的鈀碳催化劑,濾液用無水乙酸乙酯萃取,每次加入20克無水乙酸乙酯,共萃取3次。合并萃取液,分出油層,然后用去離子水水洗至中性,加入5克無水氯化鈣干燥5小時,最后過濾,于55℃,真空度為20mmhg旋蒸至恒重,得到產物1,1′-甲基苯基硅甲撐-2,2′-雙(二甲基羥基硅基)雙碳硼烷。
2.合成1,3,5-三甲基-1,3,5-三[β-(二甲基氯硅基)乙基]環三硅氮烷
在100ml的三口燒瓶中先加入17.35克除水甲苯溶液,1.35克的三乙烯基三甲基環三硅氮烷,以及0.01克卡斯特催化劑,搭好實驗裝置后,先通氮氣除去裝置內的空氣,而后通過恒壓滴液漏斗滴加配置好的1.48克二甲基氯硅烷和8.88克除水甲苯的混合溶液,滴加時間為5分鐘,在25℃的條件下反應1小時,隨后升溫至75℃反應4小時,反應液變為銀灰色,得到產物1,3,5-三甲基-1,3,5-三[β-(二甲基氯硅基)乙基]環三硅氮烷,直接進入下一步反應。
3.縮聚反應
在100ml的燒杯中加入13.00克的無水甲苯,3.72克1,1′-甲基苯基硅甲撐-2,2′-雙(二甲基羥基硅基)雙碳硼烷,以及1.05克的三乙胺,混合均勻,備用。
在上述步驟2得到的1,3,5-三甲基-1,3,5-三[β-(二甲基氯硅基)乙基]環三硅氮烷中,通過恒壓滴液漏斗滴加配制好的碳硼烷單體溶液,滴加時間為15分鐘,在室溫下反應30分鐘后,升溫至50℃,反應時間為18小時。
反應結束后,自然冷卻至室溫,抽濾除去不溶的三乙胺鹽酸鹽,濾液用無水乙醚萃取,分液,然后用去離子水水洗至中性,5克無水硫酸鎂干燥5小時,最后過濾,在55℃、真空度為20mmhg下旋蒸至恒重,得到產物,一種新型的硅基碳硼烷聚合物。產物分子結構式如下:
m和n分別為5~12的整數
4.涂層處理
將碳硼烷陶瓷前驅體溶解在二氯乙烷中,配制成質量濃度為0.12g/毫升的溶液,備用。將t-700碳纖維在氮氣保護和400℃的條件下烘烤2小時,再在質量濃度為5%的硝酸溶液中浸泡1小時,然后以去離子水淋洗至中性。再將處理好的碳纖維浸泡在碳硼烷陶瓷前驅體的二氯乙烷溶液中,室溫自然風干。重復處理5次得到有5層涂層的碳纖維材料。碳纖維在空氣的氛圍中加熱至1000℃質量保留率達92.15%,碳纖維處理后耐熱性能非常優異。
- 還沒有人留言評論。精彩留言會獲得點贊!