一種溶劑黃124的合成方法與流程
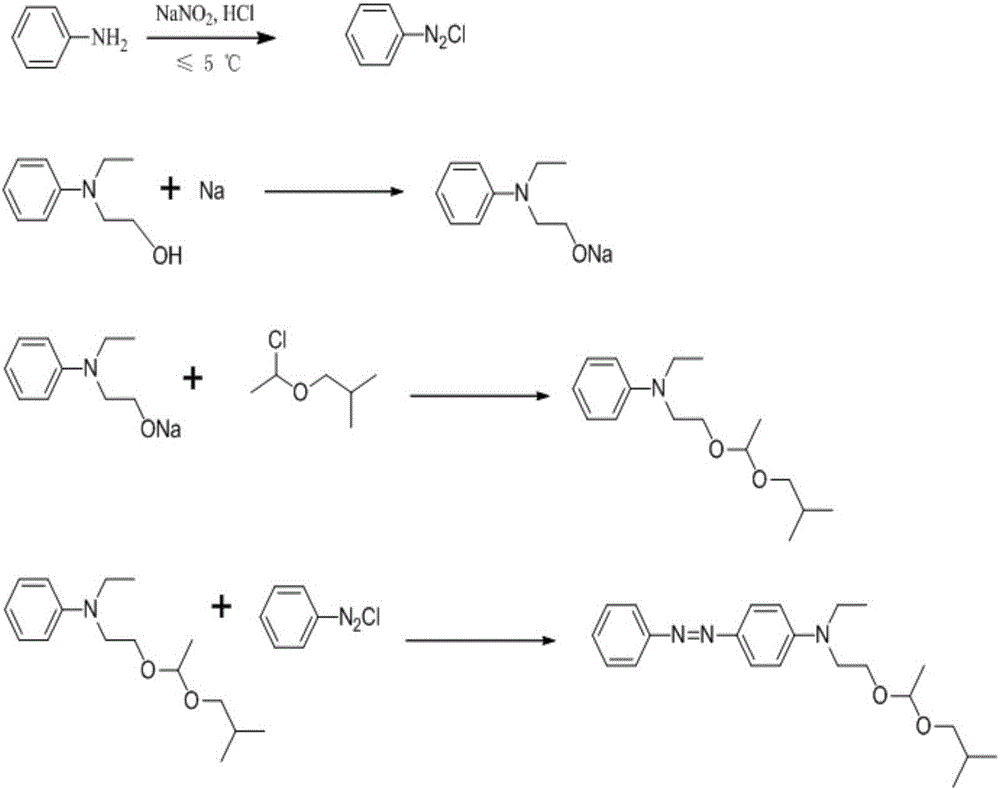
本發(fā)明涉及溶劑染料合成領(lǐng)域,具體涉及一種溶劑黃124的合成方法。
背景技術(shù):
:染料是指在一定介質(zhì)中,能使纖維或其他物質(zhì)牢固著色的化合物。目前,染料已不只限于紡織物的染色和印花,在它在油漆、塑料、紙張、皮革、光電通訊、食品等許多部門得以應(yīng)用。而溶劑染料是以能溶于有機溶劑為特征的一類染料。在這類染料結(jié)構(gòu)中不含或少含親水性基團。為了增大在溶劑中的溶解度常在染料結(jié)構(gòu)中引入長碳鏈的烷基物,使染料具有電中性,在非纖維著色方面應(yīng)用廣泛。溶劑黃124,是一個偶氮結(jié)構(gòu)的染料,其中含有不對稱的縮醛結(jié)構(gòu),是一種性能優(yōu)良的溶劑染料,主要用于塑料制品的作色,亦可經(jīng)處理后作為分散染料用于滌綸及其混紡織物的染色。由于工藝條件的限制,目前其市售產(chǎn)品主含量偏低,質(zhì)量不穩(wěn)定,同時由于生產(chǎn)工藝收率較低造成單價較高,使其應(yīng)用受到一定限制。專利號為US4011209的美國專利,公開了一種溶劑黃124的合成路線,以苯胺為原料,重氮化得到氯化重氮基苯后,經(jīng)耦合反應(yīng)后,再在三氯化硼的催化下進行縮醛反應(yīng)制備得到溶劑黃124,該反應(yīng)路線合成收率較低,副產(chǎn)物較多,同時收率較低。技術(shù)實現(xiàn)要素:為了解決上述的技術(shù)問題,本發(fā)明通過采用苯胺為原料經(jīng)重氮化后得到氯化重氮基苯,將N-乙基-N-羥乙基苯胺和經(jīng)處理后的金屬鈉顆粒反應(yīng)得到醇鈉,該醇鈉經(jīng)縮醛反應(yīng)后,再和氯化重氮基苯進行耦合反應(yīng),制備得溶劑黃124,該反應(yīng)路線合成收率高,制備出的產(chǎn)物純度高。本發(fā)明采用如下技術(shù)方案:一種溶劑黃124的合成方法,包括以下步驟:1)氯化重氮基苯的制備在反應(yīng)容器中加入苯胺、蒸餾水和濃硫酸,加熱溶解,迅速冷卻至5℃,緩慢滴加濃度為30%的亞硝酸鈉溶液,在溫度≤10℃,pH≤2下反應(yīng)40-60min,TLC檢測反應(yīng)終點;反應(yīng)結(jié)束后,加入尿素分解過量的亞硝酸鈉,得絳紅色重氮鹽溶液,用濃鹽酸調(diào)節(jié)該溶液pH至2.0-3.0,待用;2)N-乙基-N-羥乙基苯胺鈉鹽的制備將金屬鈉表面的煤油擦干后,切割成直徑為0.5-1cm的顆粒,置入裝有N-乙基-N-羥乙基苯胺的四氫呋喃溶液中,加熱回流2-6h,降至室溫,待用;3)N-乙基-N-(2-(1-異丁氧基乙氧基)乙基)苯胺的制備往裝有步驟2)待用的N-乙基-N-羥乙基苯胺鈉鹽的反應(yīng)容器中加入α-氯乙基異丁基醚,以四氫呋喃為溶液,加熱回流4-5h,反應(yīng)完畢降至室溫,真空旋干四氫呋喃,再加入20%的氯化鈉溶液,用乙酸乙酯萃取,收集乙酸乙酯層,回收至干得到油狀物,加入丙酮溶液,待用;4)溶劑黃124的合成將步驟3)待用的N-乙基-N-(2-(1-異丁氧基乙氧基)乙基)苯胺的丙酮溶液冷卻至3℃,加入有機堿調(diào)至pH=7.0~9.5,在5~10℃的條件下,在30min內(nèi)滴入步驟1)制得的氯化重氮基苯溶液,攪拌2h。鹽酸溶液調(diào)至pH=2.0-5.0,60℃加熱15min后冷卻至室溫。加入乙酸乙酯萃取,收集的乙酸乙酯層,經(jīng)飽和食鹽水洗滌,無水硫酸鎂干燥后、過濾,真空旋干溶劑,得到溶劑黃124。優(yōu)選的,所述步驟1)中反應(yīng)條件為在溫度≤5℃,pH≤1下進行。進一步的,所述步驟1)中加入的亞硝酸鈉總量與苯胺比例,按照摩爾比,亞硝酸鈉:苯胺=1.2-1.4:1。優(yōu)選的,所述步驟2)中反應(yīng)時間為4-5h。優(yōu)選的,所述步驟3)中真空旋干四氫呋喃的條件為:在真空度為0.07-0.09MPa,溫度為40-50℃下回收。進一步的,所述步驟2)-步驟3)中,所使用的金屬鈉顆粒、N-乙基-N-羥乙基苯胺、α-氯乙基異丁基醚,按照摩爾比,金屬鈉顆粒:N-乙基-N-羥乙基苯胺:α-氯乙基異丁基醚=1:1.2-1.3:1.1-1.2。優(yōu)選的,所述步驟4)中,所述的有機堿為三乙胺、三正丁胺、DBU中的任一種。優(yōu)選的,所述步驟4)中,有機堿調(diào)至pH=7.5-8.5。優(yōu)選的,所述步驟4)中,鹽酸溶液調(diào)至pH=4.0-4.5。與現(xiàn)有技術(shù)比較,本發(fā)明具有以下優(yōu)點:1、重氮化反應(yīng)穩(wěn)定,而且收率高,得到的氯化重氮基苯純度高,為耦合反應(yīng)提供了較好的基礎(chǔ)。2、現(xiàn)有技術(shù)是先進行耦合反應(yīng),然后再進行縮醛反應(yīng),該縮醛反應(yīng)條件是在酸性下進行的,會使得溶劑黃124的縮醛結(jié)構(gòu)發(fā)生分解,易生成雜質(zhì),且該雜質(zhì)不易分離;而本發(fā)明是先進行的縮醛反應(yīng),再在堿性條件下進行耦合反應(yīng),避免了溶劑黃124在酸性條件下發(fā)生分解的過程,同時本發(fā)明的選擇性好,生成的雜質(zhì)較少,從而收率高,產(chǎn)品純度高。3、本發(fā)明合成過程中,反應(yīng)條件溫和,得到的中間體純度較好,(略經(jīng)后處理)可以直接用于進行下一步反應(yīng),為工業(yè)化的連續(xù)生產(chǎn)提供了技術(shù)支持。經(jīng)本法合成的溶劑黃124的總收率達到68%,大大高于文獻報道方法合成的總收率。有較高的應(yīng)用價值,可以大幅度降低生產(chǎn)成本,提高產(chǎn)率和經(jīng)濟效益。附圖說明圖1為本發(fā)明合成方法涉及的反應(yīng)方程式圖2為國外標樣溶劑黃124的HPLC圖譜(450nm和320nm的條件下)圖3為本發(fā)明實施例1所制備出的溶劑黃124的HPLC圖譜(450nm和320nm的條件下)圖4為國外標樣溶劑黃124的紫外-可見吸收光譜圖5為本發(fā)明實施例1所制備出的溶劑黃124的的紫外-可見吸收光譜具體實施方式以下結(jié)合實施例及附圖對本發(fā)明的技術(shù)方案作進一步描述,但要求保護的范圍并不局限于所述。實施例1一種溶劑黃124的合成方法,包括以下步驟:1)氯化重氮基苯的制備在反應(yīng)容器中加入10g苯胺(0.1mol)、50ml蒸餾水和10ml濃硫酸,加熱溶解,迅速冷卻至5℃,緩慢滴加濃度為30%的亞硝酸鈉溶液(總重為27.5g),在溫度≤10℃,pH≤2下反應(yīng)40min,TLC檢測反應(yīng)終點;反應(yīng)結(jié)束后,加入2g尿素分解過量的亞硝酸鈉,得絳紅色重氮鹽溶液,用濃鹽酸調(diào)節(jié)該溶液pH至2.0-3.0,待用;2)N-乙基-N-羥乙基苯胺鈉鹽的制備將金屬鈉表面的煤油擦干后,切割成直徑為0.5-1cm的顆粒,將28.43g的N-乙基-N-羥乙基苯胺(0.17mol)和切割好的3.25g金屬鈉顆粒(0.14mol)及200ml四氫呋喃加入反應(yīng)容器后,加熱回流2h,降至室溫,待用;3)N-乙基-N-(2-(1-異丁氧基乙氧基)乙基)苯胺的制備往裝有步驟2)待用的N-乙基-N-羥乙基苯胺鈉鹽的反應(yīng)容器中加入21.50g的α-氯乙基異丁基醚(0.15mol),加入100ml四氫呋喃,加熱回流4.5h,反應(yīng)完畢降至室溫,在真空度為0.07-0.09MPa,溫度為40-50℃下旋干四氫呋喃,再加入100ml的20%的氯化鈉溶液,用乙酸乙酯溶液萃取4次,每次用量為50ml,收集乙酸乙酯層,回收至干得到油狀物,加入200ml的丙酮溶液,待用。4)溶劑黃124的合成將步驟3)待用的N-乙基-N-(2-(1-異丁氧基乙氧基)乙基)苯胺的丙酮溶液冷卻至3℃,加入三乙胺調(diào)至pH=7.0~7.5,在5~10℃的條件下,在30min內(nèi)滴入步驟1)制得的氯化重氮基苯溶液,攪拌2h。鹽酸溶液調(diào)至pH=2.0-3.0,60℃加熱15min后冷卻至室溫。加入乙酸乙酯萃取,再經(jīng)飽和食鹽水洗滌,30g無水硫酸鎂干燥、過濾,真空旋干溶劑,得到溶劑黃124。實施例2一種溶劑黃124的合成方法,包括以下步驟:1)氯化重氮基苯的制備在反應(yīng)容器中加入10g苯胺(0.1mol)、50ml蒸餾水和10ml濃硫酸,加熱溶解,迅速冷卻至5℃,緩慢滴加濃度為30%的亞硝酸鈉溶液(總重為32.2g),在溫度≤5℃,pH≤1下反應(yīng)50min,TLC檢測反應(yīng)終點;反應(yīng)結(jié)束后,加入2g尿素分解過量的亞硝酸鈉,得絳紅色重氮鹽溶液,用濃鹽酸調(diào)節(jié)該溶液pH至2.0-3.0,待用;2)N-乙基-N-羥乙基苯胺鈉鹽的制備將金屬鈉表面的煤油擦干后,切割成直徑為0.5-1cm的顆粒,將30g的N-乙基-N-羥乙基苯胺(0.18mol)和切割好的3.25g金屬鈉顆粒(0.14mol)及200ml四氫呋喃加入反應(yīng)容器后,加熱回流4h,降至室溫,待用;3)N-乙基-N-(2-(1-異丁氧基乙氧基)乙基)苯胺的制備往裝有步驟2)待用的N-乙基-N-羥乙基苯胺鈉鹽的反應(yīng)容器中加入22.50g的α-氯乙基異丁基醚(0.16mol),加入100ml四氫呋喃,加熱回流4h,反應(yīng)完畢降至室溫,在真空度為0.07-0.09MPa,溫度為40-50℃下旋干四氫呋喃,再加入100ml的20%的氯化鈉溶液,用乙酸乙酯溶液萃取4次,每次用量為50ml,收集乙酸乙酯層,回收至干得到油狀物,加入200ml的丙酮溶液,待用。4)溶劑黃124的合成將步驟3)待用的N-乙基-N-(2-(1-異丁氧基乙氧基)乙基)苯胺的丙酮溶液冷卻至3℃,加入三乙胺調(diào)至pH=7.5~8.5,在5~10℃的條件下,在30min內(nèi)滴入步驟1)制得的氯化重氮基苯溶液,攪拌2h。鹽酸溶液調(diào)至pH=3.5-4.5,60℃加熱15min后冷卻至室溫。加入乙酸乙酯萃取,再經(jīng)飽和食鹽水洗滌,30g無水硫酸鎂干燥、過濾,真空旋干溶劑,得到溶劑黃124。實施例3一種溶劑黃124的合成方法,包括以下步驟:1)氯化重氮基苯的制備在反應(yīng)容器中加入10g苯胺(0.1mol)、50ml蒸餾水和10ml濃硫酸,加熱溶解,迅速冷卻至5℃,緩慢滴加濃度為30%的亞硝酸鈉溶液(總重為30g),在溫度≤10℃,pH≤1下反應(yīng)60min,TLC檢測反應(yīng)終點;反應(yīng)結(jié)束后,加入2g尿素分解過量的亞硝酸鈉,得絳紅色重氮鹽溶液,用濃鹽酸調(diào)節(jié)該溶液pH至2.0-3.0,待用;2)N-乙基-N-羥乙基苯胺鈉鹽的制備將金屬鈉表面的煤油擦干后,切割成直徑為0.5-1cm的顆粒,將31.30g的N-乙基-N-羥乙基苯胺(0.19mol)和切割好的3.25g金屬鈉顆粒(0.14mol)及200ml四氫呋喃加入反應(yīng)容器后,加熱回流6h,降至室溫,待用;3)N-乙基-N-(2-(1-異丁氧基乙氧基)乙基)苯胺的制備往裝有步驟2)待用的N-乙基-N-羥乙基苯胺鈉鹽的反應(yīng)容器中加入22.50g的α-氯乙基異丁基醚(0.17mol),加入100ml四氫呋喃,,加熱回流4.5h,反應(yīng)完畢降至室溫,在真空度為0.07-0.09MPa,溫度為40-50℃下旋干四氫呋喃,再加入100ml的20%的氯化鈉溶液,用乙酸乙酯溶液萃取4次,每次用量為50ml,收集乙酸乙酯層,回收至干得到油狀物,加入200ml的丙酮溶液,待用。4)溶劑黃124的合成將步驟3)待用的N-乙基-N-(2-(1-異丁氧基乙氧基)乙基)苯胺的丙酮溶液冷卻至3℃,加入三乙胺調(diào)至pH=8.5~9.5,在5~10℃的條件下,在30min內(nèi)滴入步驟1)制得的氯化重氮基苯溶液,攪拌2h。鹽酸溶液調(diào)至pH=4.5-5.0,60℃加熱15min后冷卻至室溫。加入乙酸乙酯萃取,再經(jīng)飽和食鹽水洗滌,30g無水硫酸鎂干燥、過濾,真空旋干溶劑,得到溶劑黃124。實施例4一種溶劑黃124的合成方法,包括以下步驟:1)氯化重氮基苯的制備在反應(yīng)容器中加入10g苯胺(0.1mol)、50ml蒸餾水和10ml濃硫酸,加熱溶解,迅速冷卻至5℃,緩慢滴加濃度為30%的亞硝酸鈉溶液(總重為27.5g),在溫度≤5℃,pH≤2下反應(yīng)60min,TLC檢測反應(yīng)終點;反應(yīng)結(jié)束后,加入2g尿素分解過量的亞硝酸鈉,得絳紅色重氮鹽溶液,用濃鹽酸調(diào)節(jié)該溶液pH至2.0-3.0,待用;2)N-乙基-N-羥乙基苯胺鈉鹽的制備將金屬鈉表面的煤油擦干后,切割成直徑為0.5-1cm的顆粒,將28.43g的N-乙基-N-羥乙基苯胺(0.17mol)和切割好的3.25g金屬鈉顆粒(0.14mol)及200ml四氫呋喃加入反應(yīng)容器后,加熱回流5h,降至室溫,待用;3)N-乙基-N-(2-(1-異丁氧基乙氧基)乙基)苯胺的制備往裝有步驟2)待用的N-乙基-N-羥乙基苯胺鈉鹽的反應(yīng)容器中加入22.50g的α-氯乙基異丁基醚(0.16mol),加入100ml四氫呋喃,加熱回流4h,反應(yīng)完畢降至室溫,在真空度為0.07-0.09MPa,溫度為40-50℃下旋干四氫呋喃,再加入100ml的20%的氯化鈉溶液,用乙酸乙酯溶液萃取4次,每次用量為50ml,收集乙酸乙酯層,回收至干得到油狀物,加入200ml的丙酮溶液,待用。4)溶劑黃124的合成將步驟3)待用的N-乙基-N-(2-(1-異丁氧基乙氧基)乙基)苯胺的丙酮溶液冷卻至3℃,加入三正丁胺調(diào)至pH=8.5~9.5,在5~10℃的條件下,在30min內(nèi)滴入步驟1)制得的氯化重氮基苯溶液,攪拌2h。鹽酸溶液調(diào)至pH=4.0-4.5,60℃加熱15min后冷卻至室溫。加入乙酸乙酯萃取,再經(jīng)飽和食鹽水洗滌,30g無水硫酸鎂干燥、過濾,真空旋干溶劑,得到溶劑黃124。實施例5一種溶劑黃124的合成方法,包括以下步驟:1)氯化重氮基苯的制備在反應(yīng)容器中加入10g苯胺(0.1mol)、50ml蒸餾水和10ml濃硫酸,加熱溶解,迅速冷卻至5℃,緩慢滴加濃度為30%的亞硝酸鈉溶液(總重為30g),在溫度≤5℃,pH≤1下反應(yīng)50min,TLC檢測反應(yīng)終點;反應(yīng)結(jié)束后,2g尿素分解過量的亞硝酸鈉,得絳紅色重氮鹽溶液,用濃鹽酸調(diào)節(jié)該溶液pH至2.0-3.0,待用;2)N-乙基-N-羥乙基苯胺鈉鹽的制備將金屬鈉表面的煤油擦干后,切割成直徑為0.5-1cm的顆粒,將31.30gg的N-乙基-N-羥乙基苯胺(0.19mol)和切割好的3.25g金屬鈉顆粒(0.14mol)及200ml四氫呋喃加入反應(yīng)容器后,加熱回流4.5h,降至室溫,待用;3)N-乙基-N-(2-(1-異丁氧基乙氧基)乙基)苯胺的制備往裝有步驟2)待用的N-乙基-N-羥乙基苯胺鈉鹽的反應(yīng)容器中加入23.50g的α-氯乙基異丁基醚(0.17mol),加入100ml四氫呋喃,加熱回流4.5h,反應(yīng)完畢降至室溫,在真空度為0.07-0.09MPa,溫度為40-50℃下旋干四氫呋喃,再加入100ml的20%的氯化鈉溶液,用乙酸乙酯溶液萃取4次,每次用量為50ml,收集乙酸乙酯層,回收至干得到油狀物,加入200ml的丙酮溶液,待用。4)溶劑黃124的合成將步驟3)待用的N-乙基-N-(2-(1-異丁氧基乙氧基)乙基)苯胺的丙酮溶液冷卻至3℃,加入三正丁胺調(diào)至pH=7.5~8.5,在5~10℃的條件下,在30min內(nèi)滴入步驟1)制得的氯化重氮基苯溶液,攪拌2h。鹽酸溶液調(diào)至pH=2.0-3.0,60℃加熱15min后冷卻至室溫。加入乙酸乙酯萃取,再經(jīng)飽和食鹽水洗滌,30g無水硫酸鎂干燥、過濾,真空旋干溶劑,得到溶劑黃124。實施例6一種溶劑黃124的合成方法,包括以下步驟:1)氯化重氮基苯的制備在反應(yīng)容器中加入10g苯胺(0.1mol)、50ml蒸餾水和10ml濃硫酸,加熱溶解,迅速冷卻至5℃,緩慢滴加濃度為30%的亞硝酸鈉溶液(總重為33.0g),在溫度≤10℃,pH≤1下反應(yīng)40min,TLC檢測反應(yīng)終點;反應(yīng)結(jié)束后,加入2g尿素分解過量的亞硝酸鈉,得絳紅色重氮鹽溶液,用濃鹽酸調(diào)節(jié)該溶液pH至2.0-3.0,待用;2)N-乙基-N-羥乙基苯胺鈉鹽的制備將金屬鈉表面的煤油擦干后,切割成直徑為0.5-1cm的顆粒,將30g的N-乙基-N-羥乙基苯胺(0.18mol)和切割好的3.25g金屬鈉顆粒(0.14mol)及200ml四氫呋喃加入反應(yīng)容器后,加熱回流6h,降至室溫,待用;3)N-乙基-N-(2-(1-異丁氧基乙氧基)乙基)苯胺的制備往裝有步驟2)待用的N-乙基-N-羥乙基苯胺鈉鹽的反應(yīng)容器中加入21.50g的α-氯乙基異丁基醚(0.15mol),加入100ml四氫呋喃,加熱回流4h,反應(yīng)完畢降至室溫,在真空度為0.07-0.09MPa,溫度為40-50℃下旋干四氫呋喃,再加入100ml的20%的氯化鈉溶液,用乙酸乙酯溶液萃取4次,每次用量為50ml,收集乙酸乙酯層,回收至干得到油狀物,加入200ml的丙酮溶液,待用。4)溶劑黃124的合成將步驟3)待用的N-乙基-N-(2-(1-異丁氧基乙氧基)乙基)苯胺的丙酮溶液冷卻至3℃,加入DBU調(diào)至pH=7.0~8.0,在5~10℃的條件下,在30min內(nèi)滴入步驟1)制得的氯化重氮基苯溶液,攪拌2h。鹽酸溶液調(diào)至pH=4.5-5.0,60℃加熱15min后冷卻至室溫。加入乙酸乙酯萃取,再經(jīng)飽和食鹽水洗滌,30g無水硫酸鎂干燥、過濾,真空旋干溶劑,得到溶劑黃124。實施例7一種溶劑黃124的合成方法,包括以下步驟:1)氯化重氮基苯的制備在反應(yīng)容器中加入10g苯胺(0.1mol)、50ml蒸餾水和10ml濃硫酸,加熱溶解,迅速冷卻至5℃,緩慢滴加濃度為30%的亞硝酸鈉溶液(總重為28g),在溫度≤10℃,pH≤2下反應(yīng)60min,TLC檢測反應(yīng)終點;反應(yīng)結(jié)束后,加入2g尿素分解過量的亞硝酸鈉,得絳紅色重氮鹽溶液,用濃鹽酸調(diào)節(jié)該溶液pH至2.0-3.0,待用;2)N-乙基-N-羥乙基苯胺鈉鹽的制備將金屬鈉表面的煤油擦干后,切割成直徑為0.5-1cm的顆粒,將28.50g的N-乙基-N-羥乙基苯胺(0.17mol)和切割好的3.25g金屬鈉顆粒(0.14mol)及200ml四氫呋喃加入反應(yīng)容器后,加熱回流2h,降至室溫,待用;3)N-乙基-N-(2-(1-異丁氧基乙氧基)乙基)苯胺的制備往裝有步驟2)待用的N-乙基-N-羥乙基苯胺鈉鹽的反應(yīng)容器中加入21.50的α-氯乙基異丁基醚(0.15mol),加入100ml四氫呋喃,加熱回流5h,反應(yīng)完畢降至室溫,在真空度為0.07-0.09MPa,溫度為40-50℃下旋干四氫呋喃,再加入100ml的20%的氯化鈉溶液,用乙酸乙酯溶液萃取4次,每次用量為50ml,收集乙酸乙酯層,回收至干得到油狀物,加入200ml的丙酮溶液,待用。4)溶劑黃124的合成將步驟3)待用的N-乙基-N-(2-(1-異丁氧基乙氧基)乙基)苯胺的丙酮溶液冷卻至3℃,加入DBU調(diào)至pH=8.5~9.5,在5~10℃的條件下,在30min內(nèi)滴入步驟1)制得的氯化重氮基苯溶液,攪拌2h。鹽酸溶液調(diào)至pH=2.0-3.0,60℃加熱15min后冷卻至室溫。加入乙酸乙酯萃取,再經(jīng)飽和食鹽水洗滌,30g無水硫酸鎂干燥、過濾,真空旋干溶劑,得到溶劑黃124。對實施例1-實施例7制備出的溶劑黃124的收率和質(zhì)量情況統(tǒng)計分析,同時也按照US4011209的實施例,進行溶劑黃124的制備,結(jié)果如下表所示:項目總摩爾收率(%)色譜純度(%)總雜(%)實施例161.5093.255.38實施例268.2496.792.05實施例359.3993.155.10實施例469.1797.011.68實施例568.4696.222.01實施例664.7894.303.89實施例762.6093.524.78US401120915.3085.1410.21備注:總摩爾收率,以苯胺為起始物料來進行計算。從上表的實驗結(jié)果可以看出,本發(fā)明制備出的溶劑黃124,總摩爾收率高,而且色譜純度高,總雜較低;同時在步驟1)的制備過程中發(fā)現(xiàn),制備得到的氯化重氮基苯溶液,在冷藏條件下儲存,1-2天內(nèi)分解出的雜質(zhì)較少,比較穩(wěn)定,而相比專利US4011209制備出的氯化重氮基苯溶液,在相同的條件下儲存,在4-6h后,較易分解,生產(chǎn)較多的雜質(zhì)。同時對比本發(fā)明實施例和專利US4011209的實施例,發(fā)明本發(fā)明實施例所制備出的溶劑黃124,在塑料制品的作色方面,鮮艷度和色牢度遠優(yōu)于專利US4011209的實施例在在塑料制品的作色方面的效果。當前第1頁1 2 3
相關(guān)技術(shù)
網(wǎng)友詢問留言
已有0條留言
- 還沒有人留言評論。精彩留言會獲得點贊!
1