一種含有結核病變態反應原CE的藥物組合物的制作方法
本發明屬于生物
技術領域:
,具體涉及一種含有結核病變態反應原ce的藥物組合物及其制備方法和用途。
背景技術:
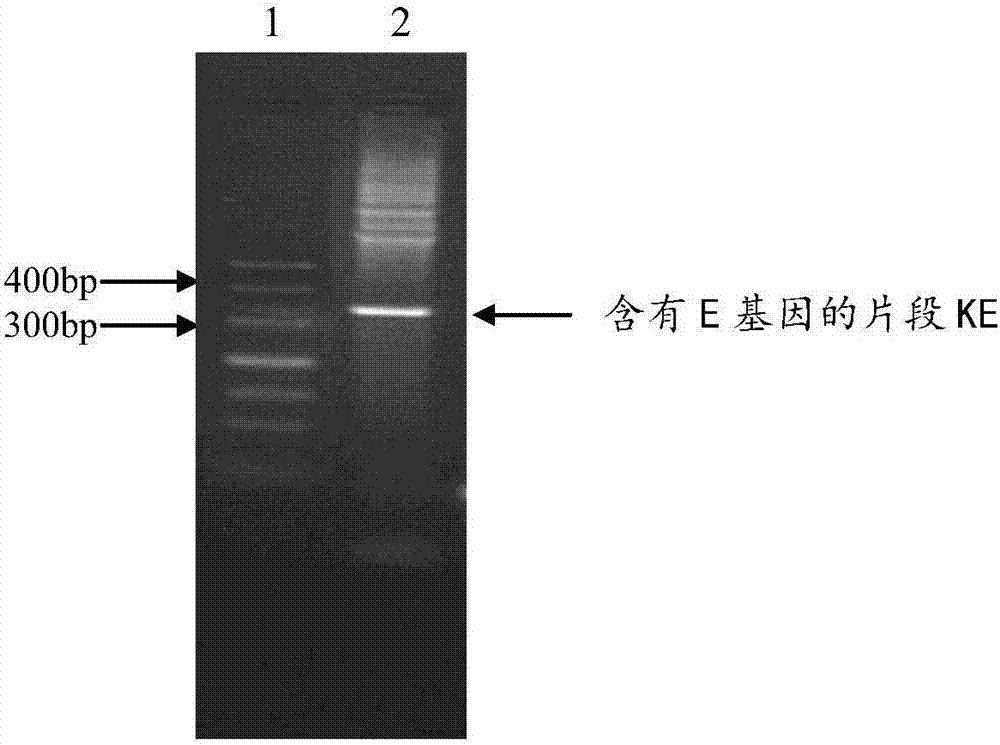
:在全世界結核病依然是危害人類健康的主要傳染病之一,是當前急需解決的重要公共健康難題之一。我國的結核病疫情和耐藥情況都相當嚴重,是全球22個結核病高負擔國家之一,結核病人數位居世界第二,僅次于印度。2010年第五次全國結核病流行病學抽樣調查初步結果表明,現有肺結核病人500-600萬,其中傳染性肺結核病人86萬;全國肺結核發病人數和死亡人數位于各種法定傳染病的首位。結核病是通過呼吸道傳播的傳染性很強的疾病,我國約有三分之一人口感染了結核菌,其中5%可能早期發病,5%在其一生中的任何時候可能發病。因此,結核高危人群的主動發現、動態監測和預防治療可有效減少肺結核的發病率,結核病的早期診斷、發現傳染源、有效化療對于控制結核病及結核菌傳播都有極其重要的意義。目前臨床上常規應用的結核病診斷方法中,用于檢測排菌肺結核患者的臨床標本涂片的靈敏度低,陽性率只有20-30%;傳統的羅氏培養的陽性率也只有30%左右,而且需4-8周時間;國際認可的分枝桿菌快速培養系統一般也需2周以上,而且試劑昂貴,檢測成本高而難以普及。用于檢測結核感染者、輔助菌陰肺結核病患者的診斷的結核菌素皮膚試驗方法的靈敏度低、特異性差,不能鑒別卡介苗接種者;血清結核抗體檢測的靈敏度和特異性不理想;近兩年開發的γ干擾素釋放試驗試劑昂貴,技術復雜,不適合于基層或現場應用。顯然目前臨床上常規應用的診斷方法不能滿足臨床醫療的需求,尤其是基層醫療單位可用的檢測方法極少。結核菌素是結核分枝桿菌培養濾液蛋白,其作用于人體會激發已致敏機體產生細胞免疫反應,激活t淋巴細胞、單核細胞和巨噬細胞,釋放大量細胞因子,并使這些細胞增殖、聚集,包裹抗原形成結節,這就是遲發型變態反應,其反應強度與細胞免疫呈平行關系。結核菌素皮膚試驗已成為目前臨床上最常用且最簡便的一種結核菌感染診斷方法,反應越強,表示結核感染可能性越大,我國一直使用硬結≥5mm為陽性,≥20mm或有水泡、壞死、淋巴結炎等為強陽性,3歲以下兒童≥15mm為強陽性的標準。用于結核病皮膚試驗的主要試劑有以下幾種:(1)舊結核菌素:不易標準化和易發生非特異性反應,目前已經很少使用。(2)人型ppd:結核分枝桿菌強毒株培養濾液的純蛋白衍化物(ppd),由于生產菌株具有很強的傳染性,國家要求該ppd的制備需在副壓車間進行,因此,北京密云的生產廠家已停產很多年,目前國內沒有廠家生產結核分枝桿菌ppd。(3)卡介菌ppd:是卡介苗菌株的培養濾液純蛋白衍化物,主要用于檢測卡介苗接種是否成功。但人型ppd停產后,臨床上用卡介菌ppd暫時替代人型ppd進行皮膚試驗,但兩者ppd成份并不完全一樣,因此,卡介菌ppd皮膚試驗輔助結核病診斷的價值尚需進一步研究。而目前國內只有成都生物制品所生產,供不應求。(4)結核分枝桿菌重組38kd蛋白:是由解放軍第三零九醫院結核病研究所研發的皮試抗原,已獲得新藥證書,轉讓給廣東東莞的公司,目前正在建廠投產,尚未用于臨床。該產品有以下特點:(1)它刺激產生的皮膚反應較輕,一般不會產生水泡和淋巴管炎等副作用;(2)它的制備無傳染性,生產不需要p3條件;(3)該蛋白存在于結核分枝桿菌復合群中,結核感染者反應強,卡介苗接種者反應弱,因此,它不能完全鑒別結核感染者和卡介苗接種者。由于ppd中含有許多分枝桿菌(包括致病性分枝桿菌、環境中非致病性分枝桿菌和卡介苗)共同的抗原,ppd皮試陽性并不能鑒別是因為結核分枝桿菌復合群感染,還是接觸環境中非結核分枝桿菌或卡介苗接種后造成的致敏。因此,ppd皮試診斷結核病的特異性差,用該方法進行結核病流行病學抽樣調查獲得的結核菌感染率是不準確的,并不能真正反映人群中結核菌感染的實際狀況。目前國內外學者通過動物模型或臨床試驗研究純化抗原、合成多肽和重組蛋白,篩選致病性結核分枝桿菌表達而卡介菌不表達的、誘導皮膚遲發型變態反應的特異抗原,以期建立新的結核皮膚診斷試劑。因此,如何開發一種價格便宜,應用簡便,無需特殊儀器,適合于基層醫療單位廣泛應用于結核高危人群的篩查,菌陰肺結核和肺外結核的輔助診斷及流行病學的調查的新的結核特異的皮膚診斷試劑是亟需解決的問題。技術實現要素:本發明提供了一種含有結核病變態反應原ce的藥物組合物,該組合物在較長的時間內和劇烈破壞條件下仍能維持藥物組合物的物理和化學穩定性,并且具有非常適合于皮下施用的ph、粘度和滲透壓。本發明提供了一種含有結核病變態反應原ce的藥物組合物,其中包括:a.結核病變態反應原ce;b.至少一種結構保護劑;c.至少一種表面活性劑;d.至少一種緩沖劑;ph為4.0-8.0。在一個優選的實施方案中,本發明所述藥物組合物包括:a.1μg/ml-10mg/ml的結核病變態反應原ce;b.1mg/ml-500mg/ml的結構保護劑;c.0.01mg/ml-5mg/ml的表面活性劑;d.1mm-100mm的緩沖液;ph為4.0-8.0。在一個優選的實施方案中,本發明所述結核病變態反應原ce是選自a)或b)的蛋白:a.seqidno.2所示的蛋白質;b.將seqidno.2所示的氨基酸序列經過一個或幾個氨基酸殘基的取代和/或缺失和/或添加且功能相同的蛋白質。本發明所述的結構保護劑選自糖類、多元醇、氨基酸或聚乙二醇類(peg)。其中所述糖類選自果糖、葡萄糖、蔗糖、海藻糖、甘露糖、乳糖、甘露糖、麥芽糖、山梨糖、葡聚糖、糊精、環糊精、羥乙基淀粉或其任意混合物;多元醇選自甘露醇、甘油、山梨糖醇、乳糖醇、麥芽糖醇、木糖醇、丙二醇或其任意混合物;氨基酸選自丙氨酸、精氨酸、天冬酰胺、天冬氨酸、半胱氨酸、谷酰胺、谷氨酸、甘氨酸、組氨酸、異亮氨酸、亮氨酸、賴氨酸、蛋氨酸、苯丙氨酸、脯氨酸、絲氨酸、蘇氨酸、色氨酸、酪氨酸、纈氨酸及其相應的鹽類或其任意混合物;聚乙二醇類選自peg3000、peg3500、peg4000或peg6000。本發明所述的表面活性劑選自聚山梨酯20、聚山梨酯21、聚山梨酯40、聚山梨酯60、聚山梨酯61、聚山梨酯65、聚山梨酯80、聚山梨酯81、聚山梨酯85或其任意混合物,以及泊洛沙姆、triton、十二烷基硫酸鈉、月桂硫酸鈉、辛基糖苷鈉、月桂基-磺基甜菜堿、聚乙二醇、聚丙二醇或其任意混合物。本發明所述的緩沖液選自乙酸/乙酸鹽、琥珀酸/琥珀酸鹽、葡萄酸/葡萄酸鹽、檸檬酸/檸檬酸鹽、抗壞血酸/抗壞血酸鹽、酒石酸/酒石酸鹽、順丁烯二酸/順丁烯二酸鹽、乳酸/乳酸鹽、碳酸/碳酸氫鹽、苯甲酸/苯甲酸鹽、咪唑、磷酸/磷酸鹽、tris/tris鹽酸鹽,以及丙氨酸、精氨酸、天冬酰胺、天冬氨酸、半胱氨酸、谷酰胺、谷氨酸、甘氨酸、組氨酸、異亮氨酸、亮氨酸、賴氨酸、蛋氨酸、苯丙氨酸、脯氨酸、絲氨酸、蘇氨酸、色氨酸、酪氨酸、纈氨酸及其相應的鹽類或混合物;或其任意混合物。本發明所述的藥物組合物可以進一步含有抗氧化劑,其中所述的抗氧化劑優選抗壞血酸、色氨酸、蛋氨酸、谷胱甘肽、硫代硫酸鈉、過氧化氫酶及螯合劑等;其中螯合劑選自由氨基多羧酸,羥基氨基羧酸、n-取代甘氨酸、檸檬酸、煙酰胺、去鐵胺和去氧膽酸鹽及其混合物,優選乙二胺四乙酸(edta)、二乙三胺五乙酸(dtpa)、次氮基三乙酸(nta)及它們的鹽。本發明中使用的螯合劑可以化合物的游離酸或者游離堿或者鹽的形式存在,也可以化合物或者相應鹽的無水、水合或者其他溶劑化物的形式存在。本發明所述的藥物組合物可以進一步含有等滲調節劑,其中所述的等滲調節劑優選氯化鈉、氯化鉀、糖類、多元醇、氨基酸;其中糖類選自果糖、葡萄糖、蔗糖、海藻糖、甘露糖、乳糖、甘露糖、麥芽糖、山梨糖、葡聚糖、糊精、環糊精、羥乙基淀粉或其任意混合物;多元醇選自甘露醇、甘油、山梨糖醇、乳糖醇、麥芽糖醇、木糖醇、乳糖醇、丙二醇或者其任意混合物;氨基酸選自丙氨酸、精氨酸、天冬酰胺、天冬氨酸、半胱氨酸、谷酰胺、谷氨酸、甘氨酸、組氨酸、異亮氨酸、亮氨酸、賴氨酸、蛋氨酸、苯丙氨酸、脯氨酸、絲氨酸、蘇氨酸、色氨酸、酪氨酸、纈氨酸及其相應的鹽類或其任意混合物。本發明的藥物組合物可以進一步含有防腐劑,所述防腐劑優選間甲酚、苯酚、苯甲醇、苯扎氯銨、苯氧乙醇和對羥基苯甲酸甲酯的一種或多種。在一個優選的實施方案中,本發明所述藥物組合物包括:a.5μg/ml-1mg/ml的結核病變態反應原ce,b.20mg/ml-200mg/ml的二糖或多元醇或其混合物,c.0.05mg/ml-1mg/ml的聚山梨酯20,d.5mm-40mm檸檬酸/檸檬酸鹽緩沖液,ph為6.0-6.6。在一個優選的實施方案中,本發明所述藥物組合物包括:a.10μg/ml-200μg/ml的結核病變態反應原ce,b.20mg/ml-100mg/ml的海藻糖或蔗糖或甘露醇或其任意混合物c.0.1mg/ml-1mg/ml的聚山梨酯20,d.5mm-40mm檸檬酸/檸檬酸鹽緩沖液,ph為6.3。本發明選擇兩種只存在于結核分枝桿菌復合群和少數致病性分枝桿菌(如堪薩斯分枝桿菌、海水分枝桿菌、蘇爾加分枝桿菌)中,而不存在于所有卡介苗菌株及絕大部分環境分枝桿菌基因組中的cfp10抗原和esat6抗原,經基因工程方法構建融合表達載體pet-30a-cfp10-esat6,并在大腸桿菌工程菌中高效表達(表達產物簡稱重組融合蛋白ce),表達產物經破碎超濾濃縮,經離子交換deae-ff,ab-ff,和q-hp純化后,得到sec-hplc單體純度>99%、rp-hplc純度>95%的結核病變態反應原ce。由于選擇的cfp10和esat6抗原是中國人群的優勢抗原融合表達,含有較多的t細胞抗原決定簇,有較強的免疫原性,能特異地誘導不同結核感染人群產生較強的遲發型變態反應,而使檢測的靈敏度提高,可鑒別結核感染者和卡介苗接種者,這對于廣泛接種卡介苗的國家是非常有益的。本發明所述的結核病變態反應原ce的編碼基因和氨基酸序列如下:編碼基因:5’-catatggcagagatgaagactgatgcagctactctggcacaagaagcaggtaacttcgaacgtatctctggtgacctgaaaacccagatcgatcaggttgaatctactgcaggttctctgcagggtcaatggcgtggtgctgcaggtactgctgcacaagctgcagttgtacgtttccaagaagcagccaataagcagaagcaggaactggatgaaatctctactaacattcgtcaggcaggtgttcaatactctcgtgctgatgaagagcaacagcaagcactgagctctcaaatgggtttcggtggtggcggatctggtggcggtatgaccgaacagcagtggaacttcgcaggtatcgaagctgcagcttctgctattcagggtaacgttacctctatccactctctgctggatgaaggtaaacagtctctgaccaaactggcagctgcatggggtggttctggttctgaagcttaccagggtgttcagcagaaatgggatgctaccgcaaccgaactgaacaacgcactgcagaacctggctcgtaccatctctgaagctggtcaggctatggcttctaccgaaggtaacgttactggtatgttcgcttaaactcgag-3’(seqidno.1)氨基酸序列:maemktdaatlaqeagnferisgdlktqidqvestagslqgqwrgaagtaaqaavvrfqeaankqkqeldeistnirqagvqysradeeqqqalssqmgfggggsgggmteqqwnfagieaaasaiqgnvtsihslldegkqsltklaaawggsgseayqgvqqkwdatatelnnalqnlartiseagqamastegnvtgmfa(seqidno.2)本發明的藥物組合物可以為水針制劑、凍干制劑或者由凍干粉末和注射用水通過雙腔卡氏瓶(dual-chambercartridge)配制得到的制劑形式。所得藥物組合物的制劑優選通過皮下注射(s.c.)、皮內注射(i.d.)、靜脈注射(i.v.)、肌肉注射(i.m.)或其它非腸胃(parenteral)形式給藥實施。本發明的藥物組合物可用于制備診斷結核病的藥物制劑,尤其適用于鑒別結核感染者和卡介苗接種者。本發明還提供了所述藥物組合物的制備方法,包括如下步驟:a.將處方量的結構保護劑、表面活性劑、緩沖鹽溶解于適量的注射用水中,攪拌溶解,使用酸/堿調節至ph為4.0-8.0,優選ph為6.0-6.6,更優選6.3;所述的酸優選鹽酸,所述的堿優選氫氧化鈉;b.將處方量的結核病變態反應原ce加入至a步驟所得的緩沖液中,攪拌均勻;c.將b步驟所得溶液過濾除菌,分裝,加塞,軋蓋得到液體制劑;或者將b步驟所得溶液過濾除菌,分裝,半加塞,冷凍干燥,再加塞,軋蓋得到凍干粉針制劑。本發明的有益效果為:將所得高靈敏度、高度表達的結核病變態反應原ce制成藥物組合物,通過加入結構保護劑及其他輔料,改善了生物藥物的微環境,大大提高了藥物組合物的在高溫、及長期儲存時的穩定性,因此具有非常廣闊的市場應用前景。附圖說明圖1為c基因第二輪pcr擴增產物瓊脂糖凝膠電泳圖。圖2為e基因第二輪pcr擴增產物瓊脂糖凝膠電泳圖。圖3為ce基因pcr擴增產物瓊脂糖凝膠電泳圖。圖4為pet-30a(+)載體質粒及含有ce基因的片段的酶切產物及回收產物瓊脂糖凝膠電泳圖。圖5為pet-30a(+)載體質粒及重組質粒的瓊脂糖凝膠電泳圖。圖6為重組質粒酶切鑒定瓊脂糖凝膠電泳圖。圖7為重組質粒ce/pet-30a測序結果。圖8為ce/pet-30a大腸桿菌工程菌iptg誘導前后的sds-page結果。圖9為ce/pet-30a大腸桿菌工程菌表達形式鑒定的sds-page結果。圖10為ce/pet-30a大腸桿菌工程菌發酵誘導時間與表達量關系的sds-page結果。圖11為重組融合蛋白ce純化過程中deaesepharosefastflow柱純化洗脫液主峰的rp-hplc分析結果。圖12為重組融合蛋白ce純化過程中aminobutylsepharose6fastflow柱純化洗脫液主峰的rp-hplc分析結果。圖13為重組融合蛋白ce純化過程中qsepharosehighperformance柱純化洗脫液主峰的rp-hplc分析結果。圖14為不同ph條件下制劑溶液的tm。圖15為高溫試驗中不同ph處方樣品的單體相對含量。圖16為凍干試驗中不同處方樣品的單體相對含量。圖17為凍干試驗中不同處方樣品的主峰純度。圖18為高溫試驗中不同處方水針樣品的單體相對含量。圖19為高溫試驗中不同處方粉針樣品的單體相對含量。圖20為高溫試驗中不同處方水針樣品的主峰純度。圖21為高溫試驗中不同處方粉針樣品的主峰純度。圖22為復溶后不同處方樣品的單體相對含量。圖23為復溶后不同處方樣品的主峰純度(rp-hplc)。圖24為高溫試驗中不同處方樣品的單體相對含量。圖25為高溫試驗中不同處方樣品的主峰純度。具體實施方式以下通過實施例對本發明進一步進行說明。必須指出,這些實施例是用于說明本發明,而不應理解為對本發明的限制。處方工藝研究概述我們從制劑ph、表面活性劑、緩沖液、結構保護劑等幾個重要方面來研究處方組成,并進行處方及劑型的篩選和優化,確定最終的制劑處方和劑型。選用2ml中性硼硅玻璃管制注射劑瓶(肖特玻璃科技(蘇州)有限公司)和13mm注射用冷凍干燥無菌粉末用溴化丁基橡膠塞(westphamaceuticalservicessingaporepte.ltd.)作為內包裝材料。我們選擇ph范圍2.6~8.0、不同結構保護劑(甘露醇、蔗糖、海藻糖、鹽酸精氨酸、甘氨酸)及表面活性劑聚山梨酯20,進行本品的處方研究并最終確認用于臨床前研究和擬臨床研究的處方。分別配置好含有各種輔料的緩沖液,使用較高蛋白濃度的原液直接稀釋得到各種制劑。分別使用微熱差示掃描量熱技術(μcaldifferentialscanningcalorimetry,dsc)測量熱穩定性、分子排阻液相色譜法(sec-hplc)和反相液相色譜法(rp-hplc)來監控其物理和化學穩定性。實施例1、重組融合蛋白ce的編碼基因的克隆及重組融合蛋白ce的表達和純化一、兩個蛋白抗原表位融合的設計分析結核分枝桿菌cfp10和esat6的基因序列和蛋白質結構,確定兩個蛋白抗原表位融合的區域、組合和順序,根據大腸桿菌的密碼子使用頻率,選擇其中高頻率的密碼子即最佳密碼子或偏愛密碼子,去除一些稀有或利用率低的密碼子,通過同義密碼子替換原有密碼子進行優化,以便所設計的基因在大腸桿菌中高水平地表達,提高蛋白產量,使蛋白生產更有效和經濟。設計得到seqidno.1所示的序列。seqidno.1中第1-6位為限制性核酸內切酶ndei酶切識別位點5’-catatg-3’(包括起始密碼子5’-atg-3’),第4-303位為cfp10蛋白抗原表位的編碼基因(以下簡稱為c基因)序列,第304-327位為連接臂的編碼基因序列5’-ggtggtggcggatctggtggcggt-3’,第328-612位為優化后的esat6蛋白抗原表位的編碼基因(以下簡稱為e基因)序列,第613-615位為終止密碼子5’-taa-3’,第617-622位為限制性核酸內切酶xhoi酶切識別位點5’-ctcgag-3’。根據seqidno.1得到cfp10和esat6蛋白抗原表位依次連接形成的重組融合蛋白ce,cfp10蛋白的抗原表位位于重組融合蛋白ce的氨基端,esat6蛋白的抗原表位位于重組融合蛋白ce的羧基端。重組融合蛋白ce的氨基酸序列如seqidno.2所示。seqidno.2中第1-100位為cfp10蛋白抗原表位序列,第101-108位為連接臂,第109-203位為esat6蛋白抗原表位序列。重組融合蛋白ce的編碼基因(以下簡稱為ce基因)序列如seqidno.1中第4-612位所示。5’-catatggcagagatgaagactgatgcagctactctggcacaagaagcaggtaacttcgaacgtatctctggtgacctgaaaacccagatcgatcaggttgaatctactgcaggttctctgcagggtcaatggcgtggtgctgcaggtactgctgcacaagctgcagttgtacgtttccaagaagcagccaataagcagaagcaggaactggatgaaatctctactaacattcgtcaggcaggtgttcaatactctcgtgctgatgaagagcaacagcaagcactgagctctcaaatgggtttcggtggtggcggatctggtggcggtatgaccgaacagcagtggaacttcgcaggtatcgaagctgcagcttctgctattcagggtaacgttacctctatccactctctgctggatgaaggtaaacagtctctgaccaaactggcagctgcatggggtggttctggttctgaagcttaccagggtgttcagcagaaatgggatgctaccgcaaccgaactgaacaacgcactgcagaacctggctcgtaccatctctgaagctggtcaggctatggcttctaccgaaggtaacgttactggtatgttcgcttaaactcgag-3’(seqidno.1)maemktdaatlaqeagnferisgdlktqidqvestagslqgqwrgaagtaaqaavvrfqeaankqkqeldeistnirqagvqysradeeqqqalssqmgfggggsgggmteqqwnfagieaaasaiqgnvtsihslldegkqsltklaaawggsgseayqgvqqkwdatatelnnalqnlartiseagqamastegnvtgmfa(seqidno.2)二、通過基因工程技術克隆重組融合蛋白ce的編碼基因1、根據重組融合蛋白ce的編碼基因序列,設計并合成表1所示的引物,用于擴增ce基因。表1用于擴增ce基因的引物2、含有c基因的片段kc的全基因合成pcr(1)以表1中的c101f、c101r、c102f、c102r、c103f、c103r、c104f、c104r、c105f、c10bamh這10條引物互相作為引物和模板,進行第一輪pcr擴增,得到第一輪pcr擴增產物。具體原理如下:引物c101f和c101r互相作為引物和模板擴增出片段1:5’-caattccatatggcagagatgaagactgatgcagctactctggcacaagaagcaggtaacttcgaacgtatctctggtgacctgaaaacccagatc-3’;引物c102f和c102r互相作為引物和模板擴增出片段2:5’-gtgacctgaaaacccagatcgatcaggttgaatctactgcaggttctctgcagggtcaatggcgtggtgctgcaggtac-3’;引物c103f和c103r互相作為引物和模板擴增出片段3:5’-gcgtggtgctgcaggtactgctgcacaagctgcagttgtacgtttccaagaagcagccaataagcagaagcaggaactggatgaaatctctactaaca-3’;引物c104f和c104r互相作為引物和模板擴增出片段4:5’-ctggatgaaatctctactaacattcgtcaggcaggtgttcaatactctcgtgctgatgaagagcaacagcaagcactgagc-3’;引物c105f和c10bamh互相作為引物和模板擴增出片段5:5’-gcaacagcaagcactgagctctcaaatgggtttcggtggtggcggatcctaataaactcgagcgg-3’;以片段1和片段2為模板,以c101f和c102r為引物,擴增出片段1+2:5’-caattccatatggcagagatgaagactgatgcagctactctggcacaagaagcaggtaacttcgaacgtatctctggtgacctgaaaacccagatcgatcaggttgaatctactgcaggttctctgcagggtcaatggcgtggtgctgcaggtac-3’;以片段1+2和擴增片段3為模板,以c101f和c103r為引物,擴增出片段1+2+3:5’-caattccatatggcagagatgaagactgatgcagctactctggcacaagaagcaggtaacttcgaacgtatctctggtgacctgaaaacccagatcgatcaggttgaatctactgcaggttctctgcagggtcaatggcgtggtgctgcaggtactgctgcacaagctgcagttgtacgtttccaagaagcagccaataagcagaagcaggaactggatgaaatctctactaaca-3’;以擴增片段1+2+3和擴增片段4為模板,以c101f和c104r為引物,擴增出片段1+2+3+4:5’-caattccatatggcagagatgaagactgatgcagctactctggcacaagaagcaggtaacttcgaacgtatctctggtgacctgaaaacccagatcgatcaggttgaatctactgcaggttctctgcagggtcaatggcgtggtgctgcaggtactgctgcacaagctgcagttgtacgtttccaagaagcagccaataagcagaagcaggaactggatgaaatctctactaacattcgtcaggcaggtgttcaatactctcgtgctgatgaagagcaacagcaagcactgagc-3’;以擴增片段1+2+3+4和擴增片段5為模板,以c101f和c10bamh為引物,擴增出片段1+2+3+4+5(該片段含有c基因和部分連接臂):5’-caattccatatggcagagatgaagactgatgcagctactctggcacaagaagcaggtaacttcgaacgtatctctggtgacctgaaaacccagatcgatcaggttgaatctactgcaggttctctgcagggtcaatggcgtggtgctgcaggtactgctgcacaagctgcagttgtacgtttccaagaagcagccaataagcagaagcaggaactggatgaaatctctactaacattcgtcaggcaggtgttcaatactctcgtgctgatgaagagcaacagcaagcactgagctctcaaatgggtttcggtggtggcggatc-3’。第一輪pcr的反應體系:10×pcr緩沖液5μl、dntps4μl、引物c101f5μl、引物c101r0.5μl、引物c102f0.5μl、引物c102r0.5μl、引物c103f0.5μl、引物c103r0.5μl、引物c104f0.5μl、引物c104r0.5μl、引物c105f0.5μl、引物c10bamh5μl、pyrobesttmdna聚合酶1μl、無菌水26μl,總計50μl。第一輪pcr的反應條件:95℃預變性5min;94℃變性30s,50℃退火30s,72℃延伸2min,30個循環;72℃延伸10min;16℃降溫2min。(2)以1μl第一輪pcr擴增產物作為模板,用表1中的c10ff和c10bamrr為引物,進行第二輪pcr擴增,得到第二輪pcr擴增產物(即,含有c基因的片段kc),序列如下:5’-ccaattccatatggcagagatgaagactgatgcagctactctggcacaagaagcaggtaacttcgaacgtatctctggtgacctgaaaacccagatcgatcaggttgaatctactgcaggttctctgcagggtcaatggcgtggtgctgcaggtactgctgcacaagctgcagttgtacgtttccaagaagcagccaataagcagaagcaggaactggatgaaatctctactaacattcgtcaggcaggtgttcaatactctcgtgctgatgaagagcaacagcaagcactgagctctcaaatgggtttcggtggtggcggatcctaataaactcgagcgg-3’。第二輪pcr的反應體系:第一輪pcr擴增產物1μl、10×pcr緩沖液5μl、dntps4μl、引物c10ff5μl、引物c10bamrr5μl、dna聚合酶1μl、無菌水29μl,總計50μl。(3)電泳回收目的片段將步驟(2)得到的第二輪pcr擴增產物進行1%瓊脂糖凝膠電泳,結果如圖1所示。圖1中,1:dna分子量標準(由上至下條帶大小依次為500bp、400bp、300bp、200bp、150bp、100bp、50bp);2:步驟(2)得到的第二輪pcr擴增產物;箭頭所指為含有c基因的片段kc。在長波紫外線照射下,用干凈的手術刀片在膠上切下要回收的含有c基因的片段kc的瓊脂塊,放入無菌的離心管中。參照瓊脂糖dna回收試劑盒中的說明書回收該片段并進行測序,測序結果正確,定量后,將其貯存于-20℃備用。3、含有e基因的片段ke的全基因合成pcr(1)以表1中的e6bglf、e61f、e61r、e62f、e62r、e63f、e63r、e64f、e64r、e65f、e65r這11條引物互相作為引物和模板,進行第一輪pcr擴增,得到第一輪pcr擴增產物。具體原理如下:引物e61f和e61r互相作為引物和模板擴增出片段6:5’-aattccatatgaccgaacagcagtggaacttcgcaggtatcgaagctgcagcttctgctattcagggtaacgttacctc-3’;引物e62f和e62r互相作為引物和模板擴增出片段7:5’-tattcagggtaacgttacctctatccactctctgctggatgaaggtaaacagtctctgaccaaactggcagctgcatggggtggttctgg-3’;引物e63f和e63r互相作為引物和模板擴增出片段8:5’-ctgcatggggtggttctggttctgaagcttaccagggtgttcagcagaaatgggatgctaccgcaaccgaactgaacaacgcac-3’;引物e64f和e64r互相作為引物和模板擴增出片段9:5’-aaccgaactgaacaacgcactgcagaacctggctcgtaccatctctgaagctggtcaggctatggcttctaccgaaggtaacgttact-3’;引物e65f和e65r互相作為引物和模板擴增出片段10:5’-tctaccgaaggtaacgttactggtatgttcgcttaaactcgagcgg-3’;以擴增片段6為模板,以e6bglf和e61r為引物,擴增出片段11:5’-gcaattccatatgagatctggtggcggtatgaccgaacagcagtggaacttcgcaggtatcgaagctgcagcttctgctattcagggtaacgttacctc-3’;以擴增片段11和擴增片段7為模板,以e6bglf和e62r為引物,擴增出片段11+7:5’-gcaattccatatgagatctggtggcggtatgaccgaacagcagtggaacttcgcaggtatcgaagctgcagcttctgctattcagggtaacgttacctctatccactctctgctggatgaaggtaaacagtctctgaccaaactggcagctgcatggggtggttctgg-3’;以擴增片段11+7和擴增片段8為模板,以e6bglf和e63r為引物,擴增出片段11+7+8:5’-gcaattccatatgagatctggtggcggtatgaccgaacagcagtggaacttcgcaggtatcgaagctgcagcttctgctattcagggtaacgttacctctatccactctctgctggatgaaggtaaacagtctctgaccaaactggcagctgcatggggtggttctggttctgaagcttaccagggtgttcagcagaaatgggatgctaccgcaaccgaactgaacaacgcac-3’;以擴增片段11+7+8和擴增片段9為模板,以e6bglf和e64r為引物,擴增出片段11+7+8+9:5’-gcaattccatatgagatctggtggcggtatgaccgaacagcagtggaacttcgcaggtatcgaagctgcagcttctgctattcagggtaacgttacctctatccactctctgctggatgaaggtaaacagtctctgaccaaactggcagctgcatggggtggttctggttctgaagcttaccagggtgttcagcagaaatgggatgctaccgcaaccgaactgaacaacgcactgcagaacctggctcgtaccatctctgaagctggtcaggctatggcttctaccgaaggtaacgttact-3’;以擴增片段11+7+8+9和擴增片段10為模板,以e6bglf和e65r為引物,擴增出片段11+7+8+9+10(該片段含有e基因):5’-gcaattccatatgagatctggtggcggtatgaccgaacagcagtggaacttcgcaggtatcgaagctgcagcttctgctattcagggtaacgttacctctatccactctctgctggatgaaggtaaacagtctctgaccaaactggcagctgcatggggtggttctggttctgaagcttaccagggtgttcagcagaaatgggatgctaccgcaaccgaactgaacaacgcactgcagaacctggctcgtaccatctctgaagctggtcaggctatggcttctaccgaaggtaacgttactggtatgttcgcttaaactcgagcgg-3’。第一輪pcr的反應體系:10×pcr緩沖液5μl、dntps4μl、引物e6bglf5μl、引物e61f0.5μl、引物e61r0.5μl、引物e62f0.5μl、引物e62r0.5μl、引物e63f0.5μl、引物e63r0.5μl、引物e64f0.5μl、引物e64r0.5μl、引物e65f0.5μl、引物e65r5μl、dna聚合酶1μl、無菌水26μl,總計50μl。第一輪pcr的反應條件:95℃預變性5min;94℃變性30s,50℃退火30s,72℃延伸2min,30個循環;72℃延伸10min;16℃降溫2min。(2)以1μl第一輪pcr擴增產物作為模板,用表1中的e6bglff和e65r為引物,進行第二輪pcr擴增,得到第二輪pcr擴增產物(即,含有e基因的片段ke),序列如下:5’-gcaattccatatgagatctggtggcggtatgaccgaacagcagtggaacttcgcaggtatcgaagctgcagcttctgctattcagggtaacgttacctctatccactctctgctggatgaaggtaaacagtctctgaccaaactggcagctgcatggggtggttctggttctgaagcttaccagggtgttcagcagaaatgggatgctaccgcaaccgaactgaacaacgcactgcagaacctggctcgtaccatctctgaagctggtcaggctatggcttctaccgaaggtaacgttactggtatgttcgcttaaactcgagcgg-3’。第二輪pcr的反應體系:第一輪pcr擴增產物1μl、10×pcr緩沖液5μl、dntps4μl、引物e6bglff5μl、引物e65r5μl、dna聚合酶1μl、無菌水29μl,總計50μl。(3)電泳回收目的片段:將步驟(2)得到的第二輪pcr擴增產物進行1%瓊脂糖凝膠電泳,結果如圖2所示。圖2中,1:dna分子量標準(由上至下條帶大小依次為500bp、400bp、300bp、200bp、150bp、100bp、50bp);2:步驟(2)得到的第二輪pcr擴增產物;箭頭所指為含有e基因的片段ke。在長波紫外線照射下,用干凈的手術刀片在膠上切下要回收的含有e基因的片段ke的瓊脂塊,放入無菌的離心管中。參照瓊脂糖dna回收試劑盒中的說明書回收該片段并進行測序,測序結果正確,定量后,將其貯存于-20℃備用。4、含有c基因的片段kc與含有e基因的片段ke的連接(1)用bamhi酶切步驟2得到的含有c基因的片段kc,得到酶切產物1。酶切體系如下:含有c基因的片段kc15μl、10×k緩沖液3μl、bamhi1μl、無菌水11μl,總計30μl。(2)用bglii酶切步驟3得到的含有e基因的片段ke,得到酶切產物2。酶切體系如下:含有e基因的片段ke15μl、10×h緩沖液3μl、bglii1μl、無菌水11μl,總計30μl。(3)將酶切產物1和酶切產物2用t4dna連接酶連接,獲得連接產物。連接體系如下:酶切產物110μl、酶切產物210μl、10×連接緩沖液3μl、t4dna連接酶1μl、無菌水6μl,總計30μl。(4)用步驟(3)得到的連接產物作為模板,用表1中的c101f和e65r為引物,進行pcr擴增,得到pcr擴增產物(含有ce基因的片段kce)。pcr的反應體系:連接產物1μl、10×pcr緩沖液5μl、dntps4μl、引物c101f1μl、引物e65r1μl、dna聚合酶1μl、無菌水37μl,總計50μl。含有ce基因的片段kce序列如下:5’-caattccatatggcagagatgaagactgatgcagctactctggcacaagaagcaggtaacttcgaacgtatctctggtgacctgaaaacccagatcgatcaggttgaatctactgcaggttctctgcagggtcaatggcgtggtgctgcaggtactgctgcacaagctgcagttgtacgtttccaagaagcagccaataagcagaagcaggaactggatgaaatctctactaacattcgtcaggcaggtgttcaatactctcgtgctgatgaagagcaacagcaagcactgagctctcaaatgggtttcggtggtggcggatctggtggcggtatgaccgaacagcagtggaacttcgcaggtatcgaagctgcagcttctgctattcagggtaacgttacctctatccactctctgctggatgaaggtaaacagtctctgaccaaactggcagctgcatggggtggttctggttctgaagcttaccagggtgttcagcagaaatgggatgctaccgcaaccgaactgaacaacgcactgcagaacctggctcgtaccatctctgaagctggtcaggctatggcttctaccgaaggtaacgttactggtatgttcgcttaaactcgagcgg-3’(seqidno.3)該片段也可直接人工合成。(5)電泳回收目的片段將步驟(4)得到的pcr擴增產物(含有ce基因的片段kce)進行1%瓊脂糖凝膠電泳,結果如圖3所示。圖3中,1:步驟(4)得到的pcr擴增產物(含有ce基因的片段kce);2:dna分子量標準(由上至下條帶大小依次為10000bp、7000bp、4000bp、2000bp、1000bp、500bp、250bp)。在長波紫外線照射下,用干凈的手術刀片在膠上切下要回收的含有ce基因的片段kce的瓊脂塊,放入無菌的離心管中。參照瓊脂糖dna回收試劑盒中的說明書回收該片段并進行測序,測序結果正確,定量后,貯存于-20℃備用。5、表達載體的構建將步驟4獲得的含有ce基因的片段kce克隆至pet-30a(+)(invitrogen公司產品)載體中,再將其轉化大腸桿菌bl21(de3)宿主菌(大連takara公司產品),經篩選獲得表達重組融合蛋白ce的ce/pet-30a大腸桿菌工程菌。具體步驟如下:(1)酶切將步驟4獲得的含有ce基因的片段kce和pet-30a(+)表達載體分別用ndei和xhoi進行雙酶切。兩個酶切體系分別如下:酶切體系1:含有ce基因的片段kce3μl、10×h緩沖液8μl、ndei2μl、xhoi2μl、無菌水65μl,總計80μl。酶切體系2:pet-30a(+)3μl、10×h緩沖液8μl、ndei2μl、xhoi2μl、無菌水65μl,總計80μl。將兩個酶切體系置于37℃孵育3h,分別得到含有ce基因的片段kce的酶切產物和pet-30a(+)質粒的酶切產物。(2)電泳回收目的片段將步驟(1)得到的pet-30a(+)質粒的酶切產物和含有ce基因的片段kce的酶切產物進行1%瓊脂糖凝膠電泳,在長波紫外線照射下,用干凈的手術刀片在膠上切下要回收dna的瓊脂塊,放入無菌的離心管中。參照瓊脂糖dna回收試劑盒中的說明書分別回收得到615bp的含有ce基因的片段kce的酶切回收片段和5234bp的pet-30a(+)質粒的酶切回收片段。將pet-30a(+)質粒、pet-30a(+)質粒的酶切產物、pet-30a(+)質粒的酶切回收片段、含有ce基因的片段kce、含有ce基因的片段kce的酶切產物、含有ce基因的片段kce的酶切回收片段進行1%瓊脂糖凝膠電泳,結果如圖4所示。圖4中,1:dna分子量標準;2:pet-30a(+)質粒;3:pet-30a(+)質粒的酶切產物;4:pet-30a(+)質粒的酶切回收片段;5:含有ce基因的片段kce;6:含有ce基因的片段kce的酶切產物;7:含有ce基因的片段kce的酶切回收片段。圖4表明,pet-30a(+)及含有ce基因的片段kce的酶切產物均得到回收,且純度較好。(3)基因片段連接將步驟(2)回收純化的含有ce基因的片段kce的酶切回收片段與pet-30a(+)質粒的酶切回收片段定量后,按1:1的摩爾比混合,進行如下連接反應,得到連接產物。連接反應體系(10μl):2×連接緩沖液5μl、a/pet-30a(+)質粒的酶切回收片段1μl、b/含有ce基因的片段kce的酶切回收片段1μl、t4dna連接酶1μl、無菌水補至10μl。混勻后置于16℃連接過夜,75℃滅活10min,冰浴后直接進行轉化。(4)連接產物的轉化次日取步驟(3)得到的連接產物5μl加入含有100μl大腸桿菌bl21(de3)感受態細胞的離心管中,冰浴0.5h;放入42℃水浴箱熱休克90s,迅速取出冰浴2min;加入lb培養液400μl,37℃恒溫搖床培養1h;加入x-gal60μl,iptg4μl,混勻,取出200-400μl涂布于含有50μg/ml硫酸卡那霉素的lb平板上。倒置平板,放37℃恒溫培養箱培育過夜至單菌落生長。(5)質粒的提取根據藍白斑篩選,隨機挑取2個白色單菌落,分別命名為pet-30a-ce-1菌落和pet-30a-ce-2菌落,并分別接種于5ml含有50μg/ml卡那霉素的lb液體培養基中,37℃恒溫振蕩培養過夜。根據《分子克隆》堿裂解方法,小量提取質粒,分別得到重組質粒pet-30a-ce-1和pet-30a-ce-2。將重組質粒pet-30a-ce-1和pet-30a-ce-2進行瓊脂糖凝膠電泳分析,結果如圖5所示。圖5中,1:dna分子量標準;2:載體質粒pet-30a(+);3:重組質粒pet-30a-ce-1;4:重組質粒pet-30a-ce-2。圖5表明,重組質粒pet-30a-ce-1和pet-30a-ce-2的大小均與預期相符。將重組質粒pet-30a-ce-1和pet-30a-ce-2進一步測序,測序結果表明重組質粒的大小均為5857bp。(6)重組質粒的鑒定①酶切鑒定分別以重組質粒pet-30a-ce-1和pet-30a-ce-2為模板,用ndei和xhoi進行雙酶切鑒定。將酶切產物在1%瓊脂糖凝膠中電泳,結果如圖6所示。圖6中,m:dl2000dnamarker;1:含有ce基因的片段kce的ndei+xhoi雙酶切產物;2:重組質粒pet-30a-ce-1的ndei+xhoi雙酶切產物;3:重組質粒pet-30a-ce-2的ndei+xhoi雙酶切產物。圖6表明,重組質粒的酶切片段大小與預期相符(含有ce基因的片段大小為615bp),為陽性重組質粒。將該種質粒命名為ce/pet-30a,其來源的菌命名為ce/pet-30a大腸桿菌工程菌。②序列測定挑選一個ce/pet-30a大腸桿菌工程菌克隆送上海生工生物技術有限公司進行t7通用引物正向測序,測序圖譜如圖7所示。測序結果如seqidno.4所示。seqidno.4中,第54-59位為限制性核酸內切酶ndei酶切識別位點5’-catatg-3’(包括起始密碼子5’-atg-3’),第57-356位為cfp10蛋白抗原表位的編碼基因(c基因)序列,第357-380位為連接臂的編碼基因序列5’-ggtggtggcggatctggtggcggt-3’,第381-665位為優化后的esat6蛋白抗原表位的編碼基因(e基因)序列,第666-668位為終止密碼子5’-taa-3’,第670-675位為限制性核酸內切酶xhoi酶切識別位點5’-ctcgag-3’。結果表明,ce/pet-30a質粒中的ce基因序列(即seqidno.4中第57-665位)與設計的序列(seqidno.1中第4-612位)完全一致。5’-gggcggaacattccctctagaataattttgtttaactttaagaaggagatatacatatggcagagatgaagactgatgcagctactctggcacaagaagcaggtaacttcgaacgtatctctggtgacctgaaaacccagatcgatcaggttgaatctactgcaggttctctgcagggtcaatggcgtggtgctgcaggtactgctgcacaagctgcagttgtacgtttccaagaagcagccaataagcagaagcaggaactggatgaaatctctactaacattcgtcaggcaggtgttcaatactctcgtgctgatgaagagcaacagcaagcactgagctctcaaatgggtttcggtggtggcggatctggtggcggtatgaccgaacagcagtggaacttcgcaggtatcgaagctgcagcttctgctattcagggtaacgttacctctatccactctctgctggatgaaggtaaacagtctctgaccaaactggcagctgcatggggtggttctggttctgaagcttaccagggtgttcagcagaaatgggatgctaccgcaaccgaactgaacaacgcactgcagaacctggctcgtaccatctctgaagctggtcaggctatggcttctaccgaaggtaacgttactggtatgttcgcttaaactcgagcaccaccaccaccaccactgagatc-3’(seqidno.4)三、ce/pet-30a大腸桿菌工程菌的誘導表達及鑒定1、將ce/pet-30a大腸桿菌工程菌接種于6ml含50μg/ml硫酸卡那霉素的lb培養液中,置37℃、200rpm恒溫振蕩器培養8h后,按體積百分含量1%轉種到50ml含50μg/ml硫酸卡那霉素的lb培養液中,置37℃、200rpm恒溫振蕩器培養至od600為0.6-0.9時,加入終濃度為0.5mmol/l的iptg,誘導1-4hr,得到菌液。將1×載樣緩沖液10μl加入來自40μl菌液的沉淀樣本,混懸后,置100℃沸水浴5min,于10000rpm離心5min后,取上清液10μl進行sds-page(分離膠濃度為12%,濃縮膠濃度為5%),電泳條件為:積層膠恒壓80v,分離膠恒壓120v,待溴酚藍電泳至凝膠底部,停止電泳。用考馬斯亮藍r250染色液染色6h,用脫色液脫色至條帶清晰。ce/pet-30a大腸桿菌工程菌iptg誘導前后的sds-page結果如圖8所示。圖8中,1:蛋白質分子量標準;2:ce/pet-30a大腸桿菌工程菌iptg誘導前;3:ce/pet-30a大腸桿菌工程菌iptg誘導1小時;4:ce/pet-30a大腸桿菌工程菌iptg誘導2小時;5:ce/pet-30a大腸桿菌工程菌iptg誘導3小時;6:ce/pet-30a大腸桿菌工程菌iptg誘導4小時;箭頭所指為重組融合蛋白ce的條帶。圖8表明,ce/pet-30a大腸桿菌工程菌菌體在相對分子質量21035da位置附近有濃重的表達條帶出現,iptg誘導3-4小時表達量最多。2、表達條件及表達形式鑒定將ce/pet-30a大腸桿菌工程菌接種于10ml含50μg/ml硫酸卡那霉素的lb培養液中,置37℃恒溫振蕩器過夜培養后,按體積百分含量1%轉種到2瓶50ml含50μg/ml硫酸卡那霉素的lb培養液中,置37℃恒溫振蕩器培養至od600為0.6-0.8時,加入終濃度為0.5mmol/l的iptg,分別于37℃與15℃進行誘導4hr,分別得到ce/pet-30a大腸桿菌工程菌37℃誘導后的菌液和15℃誘導后的菌液。分別將菌液中的菌體超聲破碎,離心后,得到ce/pet-30a大腸桿菌工程菌37℃誘導后的上清和沉淀以及ce/pet-30a大腸桿菌工程菌15℃誘導后的上清和沉淀,將各上清和沉淀分別進行sds-page電泳分析,部分結果如圖9所示。圖9中,1:蛋白質分子量標準(takara-d530a:從上至下條帶大小依次為97.2,66.4,44.3,29.0,20.1kda);2:ce/pet-30a大腸桿菌工程菌37℃誘導后的上清;3:ce/pet-30a大腸桿菌工程菌15℃誘導后的上清。圖9表明,ce/pet-30a大腸桿菌工程菌在37℃與15℃分泌表達的重組融合蛋白ce均存在于其菌液的上清中。四、重組融合蛋白ce的高密度、高表達發酵1、對ce/pet-30a大腸桿菌工程菌(重組菌)進行100l發酵罐的高密度發酵,在對數生長期進行iptg誘導,誘導后4h放罐。發酵過程如下:(1)種子培養以0.1%的接種量將甘油管中保存的ce/pet-30a大腸桿菌工程菌接入裝有200mllb培養基(含50μg/ml的卡那霉素)的500ml三角燒瓶中,200r/min,37℃培養8-12h至od600達到3.0-6.0,得到種子液。(2)發酵罐培養①發酵培養基和補料培養基的配方如表2所示。表2發酵培養基和補料培養基的配方②將步驟(1)得到的種子液按照2%的接種量接種于發酵培養基中進行發酵培養,培養溫度37℃、ph6.8、控制溶氧范圍在30-80%,采用指數流加補料策略(以發酵培養的培養液的糖含量小于1g/l為標準調節補料補加速度)。od600達到25-35時開始誘導表達,誘導劑iptg終濃度為0.5mm,誘導4h后終止發酵培養。采用100l發酵罐連續培養5批,結果如表3所示。表3發酵過程參數記錄2、分別取步驟1中ce/pet-30a大腸桿菌工程菌iptg誘導后1h、2h、3h和4h的發酵液和誘導前的ce/pet-30a大腸桿菌工程菌的發酵液進行菌體超聲破碎,離心分別取ce/pet-30a大腸桿菌工程菌誘導1h、2h、3h和4h的發酵液的上清和誘導前的ce/pet-30a大腸桿菌工程菌的發酵液的上清進行sds-page分析,結果如圖10所示。圖10中,1:蛋白分子量標準;2:誘導前的ce/pet-30a大腸桿菌工程菌的發酵液的上清;3-6分別代表ce/pet-30a大腸桿菌工程菌誘導1h、2h、3h和4h的發酵液的上清;箭頭所指為重組融合蛋白ce的條帶。結果表明,iptg誘導4h時,重組融合蛋白ce的表達量達到最大,目的蛋白表達量大于4g/l。五、重組融合蛋白ce的純化及鑒定1、菌體收集及破碎采用管式離心機收集ce/pet-30a大腸桿菌工程菌菌體,然后將菌體按體積比1∶5比例重懸于20mmtris緩沖液(ph8.0)中,以12000psi均質壓力破碎菌體2次。2、上清液收集采用10000rpm離心20min收集破碎后上清液。3、超濾濃縮ⅰ大腸桿菌菌體破碎后,釋放出大量雜質且料液的體積較大,不利于層析處理,因此首先采用超濾方法進行初步分離和濃縮。重組融合蛋白ce蛋白理論分子量為21,035da,因此首先選用300kd超濾膜進行透析,以截留離心殘留的微量固形物及部分高分子雜質,然后采用5kd超濾膜進行濃縮以去除小分子雜質,濃縮至3-5倍體積時用3-5倍置換液ⅰ(20mmtris,ph8.0)進行緩沖液置換。4、deaesepharosefastflow柱純化采用緩沖液c1a相(20mmtris,ph8.0)和c1b相(20mmtris,0.3mnacl,ph8.0)。用c1a相平衡陰離子交換柱deaesepharosefastflow柱(柱的內徑為200mm)2.0-4.0個柱體積,收集平衡液;將步驟3得到的濃縮液上柱,收集穿出液;采用0-70%b線性梯度洗脫,收集洗脫液,洗脫體積為10個柱體積,流速為30l/h,檢測波長為280nm。對洗脫液的主峰進行rp-hplc分析,rp-hplc分析條件為:c4分析柱(型號kromasilc4300-5c4),溫度30℃,檢測波長214nm,流速1ml/min。按表4進行梯度洗脫(其中流動相a為0.1%三氟乙酸-水溶液,流動相b為0.08%三氟乙酸-乙腈溶液),得到主峰蛋白。結果如圖11所示。表4洗脫程序5、aminobutylsepharose6fastflow柱純化采用緩沖液c2a相(20mmtris,ph8.0)和c2b相(20mmtris,0.3mnacl,ph8.0)。用c2a相平衡離子交換柱aminobutylsepharose6fastflow柱(柱的內徑為100mm)2.0-4.0個柱體積,將步驟4得到的主峰蛋白用c2a相稀釋1-2倍上柱,采用20%c2b相預洗1.5-2.0個柱體積后20%b-70%b線性梯度洗脫,收集洗脫液,洗脫體積為10個柱體積,流速為15l/h,檢測波長為280nm。對洗脫液的主峰進行rp-hplc分析,rp-hplc分析條件為:c4分析柱(型號kromasilc4300-5c4),溫度30℃,檢測波長214nm,流速1ml/min。按表5進行梯度洗脫(其中流動相a為0.1%三氟乙酸-水溶液,流動相b為0.08%三氟乙酸-乙腈溶液),得到主峰蛋白。結果如圖12所示。表5洗脫程序6、超濾濃縮ⅱ經步驟5的aminobutylsepharose6fastflow層析后收集的主峰蛋白,采用5kd超濾膜進行緩沖液置換。首先將目的蛋白濃縮3-5倍體積后,然后用置換液ⅱ(20mmtris,ph8.0溶液)置換3-5倍體積至透過液電導至2.0以下。7、qsepharosehighperformance柱純化采用緩沖液c3a相(20mmtris,ph8.0)和c3b相(20mmtris,0.3mnacl,ph8.0)。用c3a相平衡離子交換柱qsepharosehighperformance柱(柱的內徑為50mm)2.0-4.0個柱體積,收集平衡液,將步驟6得到的濃縮液上柱,收集穿出液,采用0-70%b線性梯度洗脫,收集洗脫液,洗脫體積為10個柱體積,流速為30l/h。檢測波長為280nm。對洗脫液的主峰進行rp-hplc分析,rp-hplc分析條件為:c4分析柱(型號kromasilc4300-5c4),溫度30℃,檢測波長214nm,流速1ml/min。按表6進行梯度洗脫(其中流動相a為0.1%三氟乙酸-水溶液,流動相b為0.08%三氟乙酸-乙腈溶液),收集主峰蛋白。結果如圖13所示。表6洗脫程序8、超濾濃縮ⅲ經步驟7的qsepharosehighperformance層析后收集主峰蛋白,經5kd超濾膜濃縮至1.0-1.5l,用置換液iii(5.488g/l檸檬酸鈉(二水)、0.281g/l檸檬酸(一水)、0.4g/l聚山梨酯20,ph6.3)置換12-15倍體積,控制最終濃縮液總體積為1-1.5l。按照設定體積加入終濃度94.5g/l海藻糖(二水)攪拌溶解,調節ph至6.3±0.1,最后用置換液iii(5.488g/l檸檬酸鈉(二水)、0.281g/l檸檬酸(一水)、0.4g/l聚山梨酯20,ph6.3)補齊至設定體積。9、除菌過濾將步驟8得到的超濾濃縮液經0.22μm除菌過濾器過濾于無菌pe瓶(nalgene)中即為原液。于-70±10℃避光密封保存,有效期暫定為24個月。六、重組融合蛋白ce的鑒定1、步驟五最后純化的重組融合蛋白ce的n端15個氨基酸分析結果為maemktdaatlaqea,與設計的重組融合蛋白ce(seqidno.2所示的蛋白)n端的15個氨基酸序列完全一致。2、純化的重組融合蛋白ce的質譜測定分子量為21035da,與理論計算的分子量21035da(seqidno.2所示的蛋白的分子量)非常接近。實施例2:處方ph值研究溶液ph是影響生物藥物物理化學穩定性的重要參數之一。ph可以調節蛋白質表面的電荷分布,從而影響生物藥物分子內及分子間的作用力,并直接影響蛋白質溶解度,如蛋白質在接近其等電點(pi)的ph環境中具有較小的溶解度,也可能有較低的膠體穩定性(colloidalstability)。一.處方設計通過考察不同ph值對本品制劑溶液穩定性的影響來確定最佳ph。ph范圍設計為2.6~8.0(20mm檸檬酸鹽-磷酸鹽緩沖體系),每隔0.2個ph設計1個處方,共制備28個不同ph的處方。二.處方配置分別配置好20mm的ph2.6檸檬酸/檸檬酸鹽緩沖液和20mm的ph8.0磷酸鹽緩沖液,隨后分別按照不同的比例混合配置成最終所需要的ph。各處方中結核病變態反應原ce的濃度均為200μg/ml(不含其它輔料)。三.dsc篩選蛋白融化溫度(meltingtemperature,tm)從側面反映了生物藥物在水溶液中的穩定性,tm越大說明生物分子發生變性的溫度越高,即在實際保存條件下發生構象改變(conformationalchange)的可能越低。采用dsc對28個不同ph處方溶液進行熱穩定性研究。dsc篩選結果見圖14。由圖14中可以看出:tm基本呈正態分布,ph4.4時蛋白具有最高tm(68℃),說明在該ph下重組融合蛋白ce抗原具有最佳構象穩定性。在ph3.2~6.6范圍內,樣品的tm在60℃以上,表現出較好的熱穩定性。四.影響因素試驗根據dsc高通量數據,ph過低或過高時,結核病變態反應原重組融合蛋白ce的熱穩定均較差,因此我們重點研究中間ph的樣品穩定性,結合本品的等電點pi為4.7,選擇部分樣品(ph2.6、ph3.2、ph3.8、ph4.4、ph4.6、ph4.8、ph5.0、ph5.4、ph5.6、ph6.0、ph6.6、ph7.2、ph8.0)進行影響因素試驗,sec-hplc方法考察目標蛋白(單體)相對含量的變化。高溫試驗:將篩選后的處方樣品置于40±2℃恒溫箱中,于14天后取樣進行檢測,考察單體相對含量(sec-hplc),測得單體相對含量見表7、圖15。表7高溫試驗結果注:實驗數據表示為平均值±sd。除非另有說明,重復次數n≥2,下同。各處方單體相對含量(%)為與0時比較的結果,樣品0時單體相對含量均默認為100%,下同。高溫試驗后,ph2.6~4.4和ph7.2~ph8.0的處方單體相對含量在91.7~97.9%,ph4.6~ph6.6較為穩定,處方單體相對含量均大于98.2%。五.結論dsc試驗與影響因素-高溫試驗所反映的蛋白質物理穩定性趨勢基本一致,即較低ph和較高ph時,單體含量較低、穩定性差,ph在中間范圍時單體含量較高、穩定性好。dsc試驗中,ph為4.4左右時蛋白熱力學穩定性最好,但高溫試驗中相對較高ph處方較穩定。這可能是在ph4.4左右由于和結核病變態反應原重組融合蛋白ce的等電點(pi為4.7)接近,雖然其具有較高的構象穩定性,但是由于分子基本不帶電荷,分子間的相互排斥作用較弱,即膠體穩定性較差,造成ph穩定性曲線的漂移。由上述試驗結果顯示:ph為4.4~6.6時熱穩定性(構象穩定性)效果較高,ph為6.0~7.2時單體含量結果較理想,因此ph6.0~6.6為最佳,最終確定采用ph6.3開展后續試驗。實施例3:處方結構保護劑篩選及劑型確認研究蛋白質藥物常用的結構保護劑有多元醇(如甘露醇、甘油)、糖類(如蔗糖和海藻糖)和氨基酸(如甘氨酸、精氨酸)等。上述結構保護劑的保護機制被稱為“優先排除(preferentialexclusion)”,即在水溶液中,蛋白將保護劑分子優先排除于蛋白表面之外,加入這些保護劑會增加蛋白質的化學勢能。由于蛋白質變性、去折疊后表面積增加,和輔料的相互作用增加,引起化學勢能升高更大,從而蛋白質發生構象變性的活化能增大,使蛋白質天然構象和變性構象間的平衡向天然構象轉移,從而增加了蛋白質的構象穩定性。一.處方設計處方預篩選確定本品處方的ph為6.3,蛋白含量為200μg/ml,在此基礎上,對各類保護劑進行篩選研究,處方篩選設計方案見表8。表8處方篩選設計方案處方1只含20mm檸檬酸/檸檬酸鈉緩沖液而不含任何保護劑。其余處方在處方1的基礎上分別加入不同類型的保護劑和保護劑組合(聚山梨酯20、甘露醇、蔗糖、海藻糖、鹽酸精氨酸和甘氨酸)。處方2、3、4、5和6分別含0.1mg/ml聚山梨酯20、46mg/ml甘露醇(接近等滲溶液,下同)、90mg/ml蔗糖、94.5mg/ml海藻糖、52.5mg/ml鹽酸精氨酸、18.8mg/ml甘氨酸。處方8含40mg/ml甘露醇和15mg/ml蔗糖,處方9含94.5mg/ml海藻糖和2.56mg/ml甘油,處方10含0.1mg/ml聚山梨酯20和90mg/ml蔗糖,處方11含0.1mg/ml聚山梨酯20和94.5mg/ml海藻糖,處方12含0.1mg/ml聚山梨酯20、40mg/ml甘露醇和15mg/ml蔗糖。將配置好的各處方樣品進行灌裝,每組處方共灌裝44支,每支1ml,其中22支經凍干制成凍干粉針,另22支為水針。灌裝后即進行檢測單體蛋白相對含量,作為0時檢測結果,見凍干試驗項下結果總結表10。二.凍干試驗凍干樣品使用冷凍干燥機(christ2-6dlpcplus)進行冷凍干燥,凍干工藝過程見表9。觀察凍干后各處方樣品的外觀,再對各處方樣品的凍干粉末進行復溶,檢測樣品的外觀、可見異物、單體相對含量及主峰純度。表9結核病變態反應原重組融合蛋白ce小試凍干工藝在各處方凍干樣品中加入凍干過程中丟失的水量進行復溶,復溶后檢測樣品的外觀、可見異物、單體相對含量及主峰純度,結果見表10,單體相對含量變化見圖16,主峰純度變化見圖17。表10各處方凍干前0時及凍干后復溶檢測結果復溶后各處方的外觀和可見異物檢查均合格。處方1(無保護劑)、處方3(含46mg/ml甘露醇)和處方9(含94.5mg/ml海藻糖和2.56mg/ml甘油)的單體含量損失較大,含量分別為83.1%,78.6%和82.4%;其他處方無明顯損失,說明這些輔料成分或組合物都能在不同程度上保護蛋白免受凍干過程中的物理破壞。rp-hplc主峰純度與凍干前相比基本沒有變化,均約為95%。三.影響因素試驗將各處方的凍干和水針制劑進行高溫試驗。將各處方水針及凍干粉針樣品分別放置于40±2℃恒溫箱中,第7、14天分別取樣,檢測樣品外觀、可見異物、單體相對含量(sec-hplc)以及主峰純度(rp-hplc)。樣品外觀檢測結果見表11,可見異物檢測結果見表12,單體相對含量結果見表13,主峰純度見表14;單體相對含量變化見圖18~19,主峰純度變化見圖20~21。表11高溫試驗條件下水針及凍干粉針復溶后外觀檢測結果水針:高溫(40±2℃)條件下,14天檢測期內各處方的水針樣品的外觀檢查結果均為“無色澄明液體”。凍干粉針:高溫(40±2℃)條件下,14天檢測期內各處方的粉末外觀沒有發生顯著變化,用水復溶后,只有處方1、2、11、12的外觀為無色澄明液體,其他處方均出現乳光。表12高溫試驗條件下水針及凍干粉針復溶后可見異物檢測結果水針:高溫(40±2℃)條件下,只有處方5、7、11的可見異物在7天、14天時均符合規定。凍干粉針:高溫(40±2℃)條件下,只有處方4、5、6、8、10、11、12的可見異物在7天、14天時均符合規定。表13高溫試驗條件下水針及凍干粉針復溶后單體相對含量檢測結果水針:高溫試驗(40±2℃)條件下放置7天后,所有水針處方的單體相對含量均在98.5%以上,放置14天后,不含保護劑的處方1和含有甘氨酸的處方7的單體含量比其他處方下降明顯,分別為96.6%和96.7%。凍干粉針:高溫試驗(40±2℃)條件下,經過7天和14天,各個處方的單體相對含量差異較大,但基本上都和凍干0時較接近,說明大多數的降解是由于凍干過程引起的,由于高溫引起的破壞比例較低。既含有聚山梨酯20又含有結構保護劑(蔗糖、海藻糖、甘露醇)的處方10、11、12,14天檢測期內單體相對含量均在99%以上。表14高溫試驗條件下水針及凍干粉針復溶后主峰純度檢測結果水針:高溫試驗(40±2℃)條件下,各個處方的rp-hplc主峰純度下降明顯;14天時各處方的水針主峰純度為81.9%~89.3%,其中處方11和處方12純度下降最顯著,說明高溫破壞下化學降解顯著。凍干粉針:高溫試驗(40±2℃)條件下,各處方的凍干粉針主峰純度較穩定,14天時主峰純度均在92.9%以上,且處方之間差異不顯著。四.結論上述處方研究結果表明,本品水針在高溫條件下物理穩定性較好,單體損失量較少;結核病變態反應原重組融合蛋白ce水針制劑化學降解較多,這些化學降解較難通過各種保護劑來抑制。而在凍干粉末中,各個處方具有較高的化學穩定性和物理穩定性。不同保護劑對結核病變態反應原重組融合蛋白ce在水針和凍干條件下的保護作用是不同的,而且針對物理穩定性(sec-hplc)和化學穩定性(rp-hplc)的保護作用也不同,有些在水溶液中有保護作用,有些在凍干制劑中有保護作用,而有些保護劑均有保護作用。綜合表12、13、14的數據,分別以無保護劑的處方1的實驗結果為參照,將其余11個處方中結構保護劑的保護效果進行對比,所得結果如見表15。所研究的各個輔料均有某種程度上的保護作用。表15水針制劑和凍干制劑篩選結果表注:+表示保護劑對某項指標有保護作用,—表示沒有或基本沒有保護作用而與水針相比,凍干制劑具有顯著的優越性。凍干制劑的物理穩定性和保護劑相關,聚山梨酯20和結構保護劑如甘露醇、蔗糖、海藻糖等對蛋白都有非常顯著的保護效果,且結核病變態反應原重組融合蛋白ce各凍干處方都具有很高的化學穩定性。綜合本品特性以及穩定性數據,最終將本品劑型確定為注射用無菌粉末。處方11(含94.5mg/ml海藻糖和0.1mg/ml聚山梨酯20)與處方12(含40mg/ml甘露醇,15mg/ml蔗糖和0.1mg/ml聚山梨酯20)的凍干制劑在各項試驗中均表現出很高的穩定性,且處方11略優于處方12。因此在下輪研究中進一步分析了處方11在最終制劑蛋白濃度(10μg/ml)的穩定性。另外由于聚山梨酯20對本品凍干制劑的顯著保護作用,故在下面研究中也比較了不同聚山梨酯20含量對結核病變態反應原重組融合蛋白ce穩定性的影響。實施例4:聚山梨酯20濃度研究一.處方設計本輪處方篩選研究的目的是比較最終制劑蛋白濃度(10μg/ml)條件下含有不同聚山梨酯20含量的處方11的穩定性。在處方11的基礎上,分別設計4種在10μg/ml蛋白濃度下不同濃度的聚山梨酯20(0.1mg/ml、0.4mg/ml、0.7mg/ml、1.0mg/ml)凍干處方,將其分別命名為處方11-1、11-2、11-3和11-4,處方篩選設計方案見表16。對該四個處方進行穩定性對比研究,以確定最終處方中的聚山梨酯20含量。處方中本品的蛋白濃度為10μg/ml,將配置好的各處方樣品進行灌裝,每組處方40支,每支1ml,分裝于西林瓶中。灌裝后即進行外觀、可見異物、單體含量和純度檢測,作為0時檢測結果。表16不同聚山梨酯20濃度處方組分配制(蛋白含量為10μg/ml)處方編號聚山梨酯20(mg/ml)海藻糖(mg/ml)檸檬酸/檸檬酸鹽(mm)ph處方11-10.194.5206.3處方11-20.494.5206.3處方11-30.794.5206.3處方11-41.094.5206.3二.凍干試驗將灌裝后的處方11-1、11-2、11-3、11-4樣品經凍干制成凍干粉針(相應的凍干工藝見表9),觀察凍干后各處方樣品的外觀,再對各處方樣品的凍干粉末進行復溶,檢測樣品的外觀、可見異物、單體相對含量及主峰純度。檢測結果見表17,單體相對含量變化見圖22,主峰純度變化見圖23。凍干后,各處方都形成了堅固的外形。表17不同聚山梨酯20含量處方凍干復溶后檢測結果由表17可知,在凍干樣品中加入凍干時丟失的水量復溶后,各處方的外觀和可見異物均合格。圖22顯示與凍干前相比,各處方凍干后復溶的單體含量沒有發生顯著變化。圖23顯示處方11-1的主峰純度與凍干前相比略有下降(從98.2%到97.5%),而其他處方基本沒有變化,均為約97.5%。即四個處方均能能保護結核病變態反應原ce免受凍干過程中的物理和化學破壞,處方11-2,11-3和11-4略優于處方11-1。三.影響因素試驗將上述四個處方凍干樣品分別進行高溫試驗。將四個處方凍干粉針樣品分別置于40±2℃恒溫箱中,第7、14天分別取樣。檢測樣品外觀和可見異物、單體相對含量(sec-hplc)以及主峰純度(rp-hplc)。所得檢測樣品外觀和可見異物結果見表18,單體相對含量結果見表19,主峰純度結果見表20;單體相對含量變化見圖24,主峰純度變化見圖25。表18高溫試驗條件下凍干粉針復溶后外觀及可見異物檢測結果高溫(40±2℃)條件下,14天檢測期內,所有處方樣品的外觀均為無色澄明液體,可見異物均符合規定。表19高溫試驗條件下單體相對含量檢測結果高溫(40±2℃)條件下,14天檢測期內,單體相對含量變化不顯著,表明提高聚山梨酯20含量并不能夠進一步改善結核病變態反應原重組融合蛋白ce在高溫中的穩定性。表20高溫試驗條件下主峰純度檢測結果高溫(40±2℃)條件下,14天檢測期內,處方11-1樣品的主峰純度與0時相比有一定降低,而其他3個處方無顯著差異,同樣說明0.4mg/ml以上聚山梨酯20含量能夠在一定程度上改善結核病變態反應原重組融合蛋白ce在高溫中的化學穩定性。在確保聚山梨酯20對本品有最佳保護效果的基礎上選用最低的聚山梨酯20含量,最終將聚山梨酯20的含量確定為0.4mg/ml(即處方11-2)。實施例5:穩定性研究按實施例4中的處方11-2進行了3批樣品的生產,對各項檢測指標進行檢測,各取樣品進行24個月長期穩定性試驗(5±3℃,避光儲存),然后取樣檢測,所得結果如表21所示,各項檢測指標均較為穩定,表明本品所選定生產處方和制備工藝較為穩定,質量可控,可滿足本品臨床用藥的生產、保存、運輸以及臨床使用的需要,生產處方以及制備工藝可行。表21三批中試規模成品長期穩定性檢驗結果匯總實施例6:制劑工藝過程制劑處方11-2的制備工藝如下(室溫):(1)原液的制備:按照實施例1制得的含有結核病變態反應原ce的原液,其中結核病變態反應原ce濃度為9.57mg/ml,另含5.49mg/ml檸檬酸鈉(二水)、0.28mg/ml檸檬酸(一水)、94.5mg/ml海藻糖(二水)、0.4mg/ml聚山梨酯20,ph6.3。(2)緩沖液的配置:稱取檸檬酸鈉(二水)30.20g、檸檬酸(一水)1.55g、海藻糖(二水)519.94g、聚山梨酯202.20g,加入約5.45l注射用水,攪拌溶解,然后滴加鹽酸調節ph至6.3,再加注射用水定容至5.50l。(3)稱量取原液5.96g(密度1.036g/ml),加入至緩沖液中,混勻,過濾除菌,分裝成4700瓶,半加塞,按表10的凍干過程冷凍干燥,再加塞,軋蓋,得到結核病變態反應原ce的凍干粉針制劑。實施例7:不同ph、結構保護劑、檸檬酸/檸檬酸鹽緩沖液及聚山梨酯20濃度的結核病變態反應原ce制劑的制備我們設計并配制了不同ph、結構保護劑、檸檬酸/檸檬酸鹽緩沖液濃度及聚山梨酯20濃度的結核病變態反應原ce液體制劑(表22),并進行冷凍干燥得到相應的凍干制劑。表22不同ph、蔗糖、結構保護劑、檸檬酸/檸檬酸鹽緩沖液濃度及聚山梨酯20濃度的結核病變態反應原ce制劑當前第1頁12
相關技術
網友詢問留言
已有0條留言
- 還沒有人留言評論。精彩留言會獲得點贊!
1